Mouse Visium Brain¶
Tip
You’re using the same Giotto version with which this tutorial was written.
Install Python Modules¶
To run this vignette you need to install all of the necessary Python modules.
Important
Python module installation can be done either automatically via our installation tool (from within R) (see step 2.2A) or manually (see step 2.2B).
See Part 2.2 Giotto-Specific Python Packages of our Giotto Installation section for step-by-step instructions.
Set-Up Giotto¶
library(Giotto)
Set A Working Directory¶
#results_folder = '/path/to/directory/'
results_folder = '/Volumes/Ruben_Seagate/Dropbox (Personal)/Projects/GC_lab/Ruben_Dries/190225_spatial_package/Results/Visium/Brain/201226_results//'
Set A Giotto Python Path¶
# set python path to your preferred python version path
# set python path to NULL if you want to automatically install (only the 1st time) and use the giotto miniconda environment
python_path = NULL
if(is.null(python_path)) {
installGiottoEnvironment()
}
Dataset Explanation¶
10X genomics recently launched a new platform to obtain spatial expression data using a Visium Spatial Gene Expression slide. The Visium brain data to run this tutorial can be found here.
Note
Visium Technology
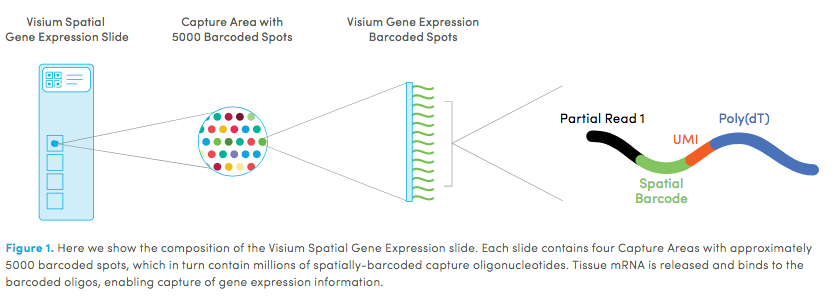
High resolution png from original tissue:
Dataset Download¶
The merFISH data to run this tutorial can be found here. Alternatively you can use the getSpatialDataset to automatically download this dataset like we do in this example.
# download data to working directory ####
# if wget is installed, set method = 'wget'
# if you run into authentication issues with wget, then add " extra = '--no-check-certificate' "
getSpatialDataset(dataset = 'cycif_PDAC', directory = results_folder, method = 'wget')
1. Giotto Global Instructions and Preparations¶
## create instructions
instrs = createGiottoInstructions(save_dir = results_folder,
save_plot = TRUE,
show_plot = FALSE)
## provide path to visium folder
#data_path = '/path/to/Brain_data/'
data_path = '/Volumes/Ruben_Seagate/Dropbox (Personal)/Projects/GC_lab/Ruben_Dries/190225_spatial_package/Data/Visium_data/Brain_data/'
2. Create Giotto Object and Process The Data¶
## directly from visium folder
visium_brain = createGiottoVisiumObject(visium_dir = data_path, expr_data = 'raw',
png_name = 'tissue_lowres_image.png',
gene_column_index = 2, instructions = instrs)
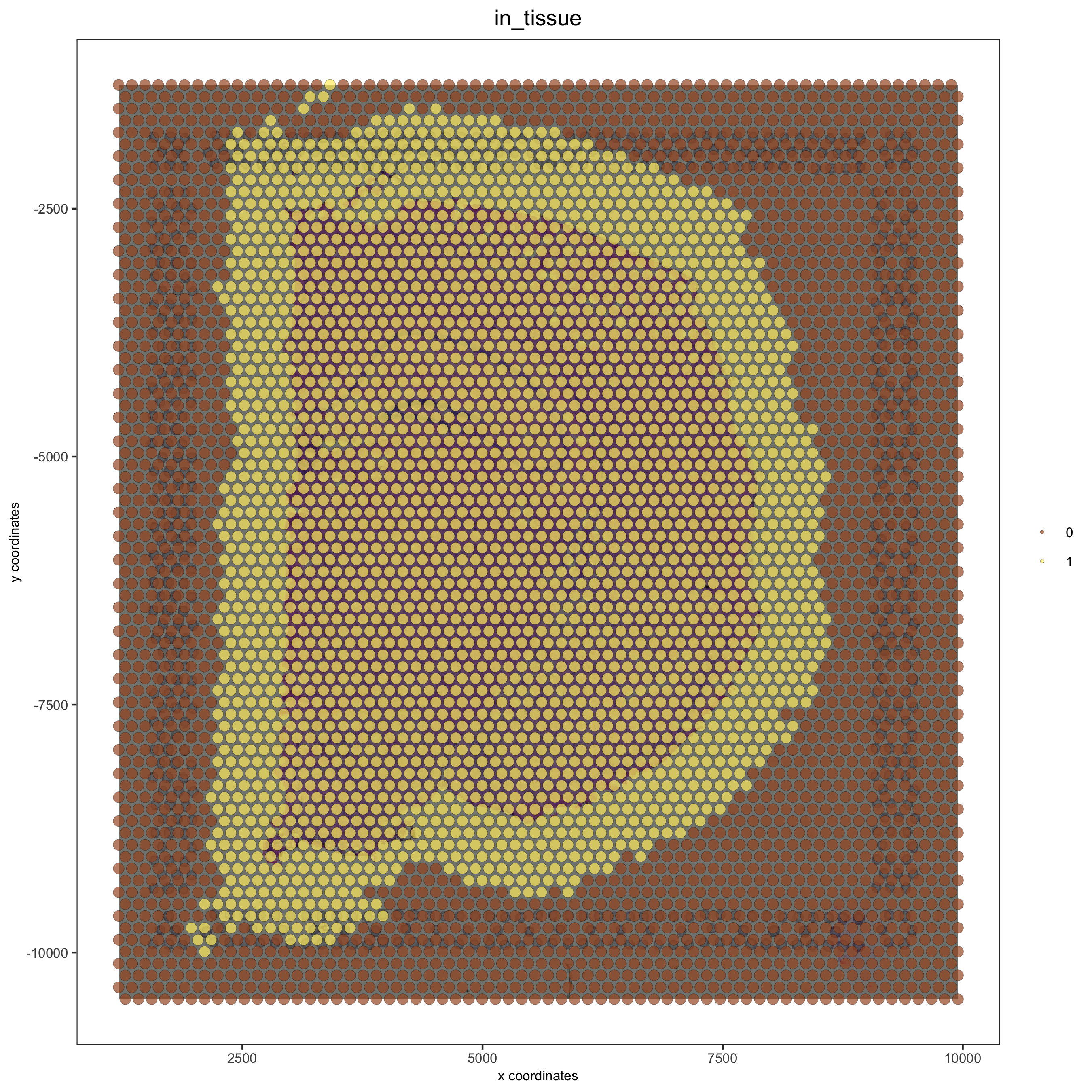
## update and align background image
# problem: image is not perfectly aligned
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,
save_param = list(save_name = '2_a_spatplot_image'))
# check name
showGiottoImageNames(visium_brain) # "image" is the default name
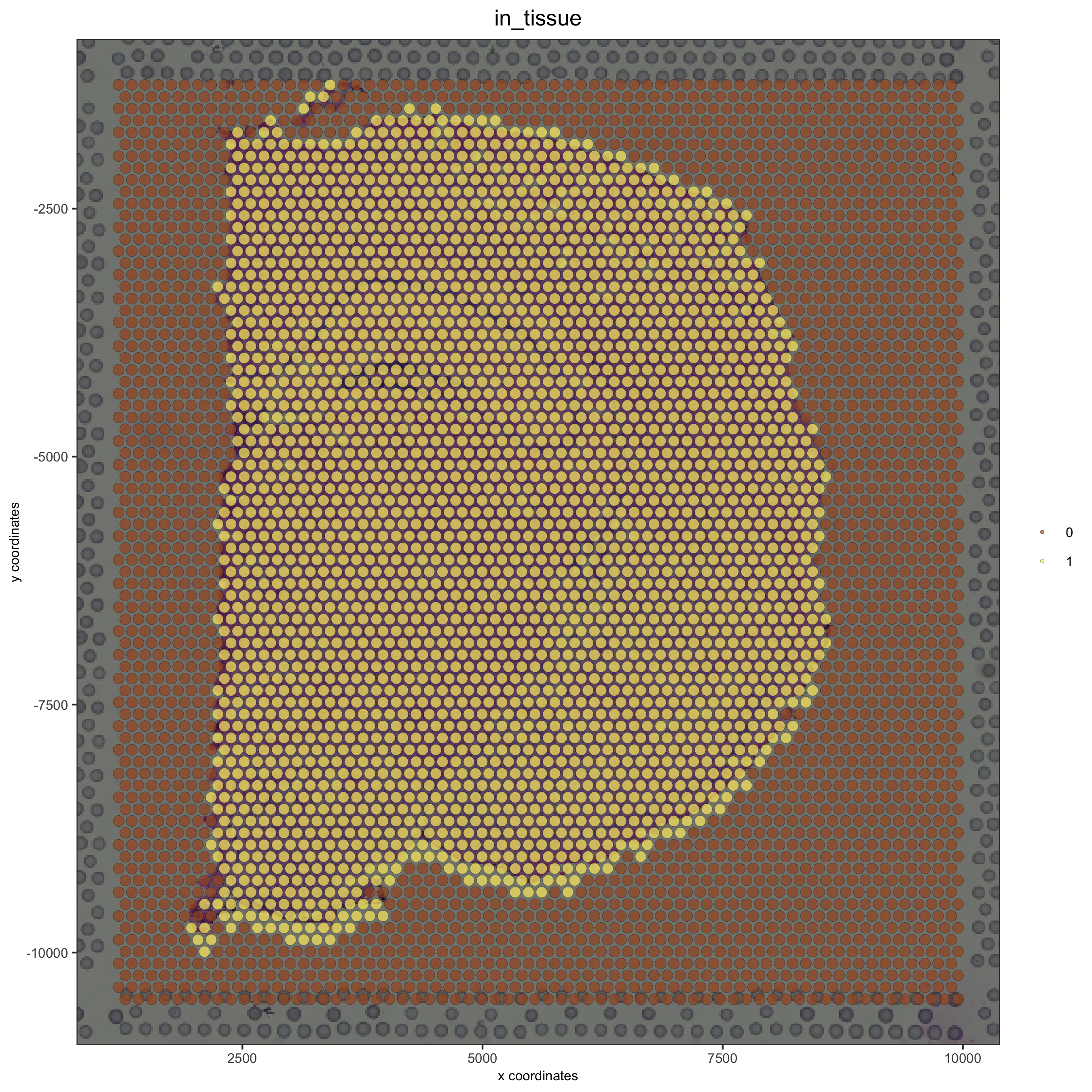
# adjust parameters to align image (iterative approach)
visium_brain = updateGiottoImage(visium_brain, image_name = 'image',
xmax_adj = 1300, xmin_adj = 1200,
ymax_adj = 1100, ymin_adj = 1000)
# now it's aligned
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,
save_param = list(save_name = '2_b_spatplot_image_adjusted'))
## check metadata
pDataDT(visium_brain)
## compare in tissue with provided jpg
spatPlot(gobject = visium_brain, cell_color = 'in_tissue', point_size = 2,
cell_color_code = c('0' = 'lightgrey', '1' = 'blue'),
save_param = list(save_name = '2_c_in_tissue'))
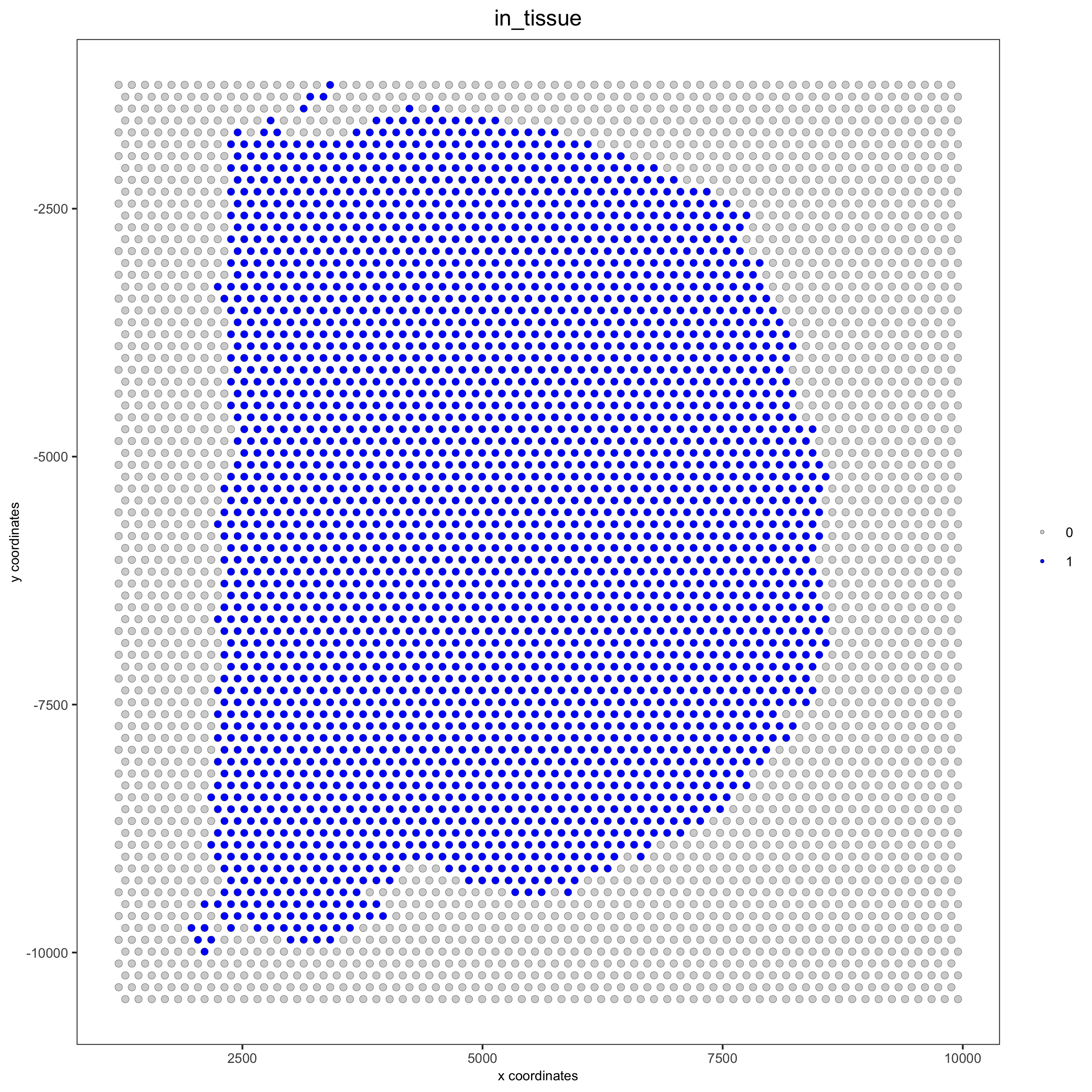
## subset on spots that were covered by tissue
metadata = pDataDT(visium_brain)
in_tissue_barcodes = metadata[in_tissue == 1]$cell_ID
visium_brain = subsetGiotto(visium_brain, cell_ids = in_tissue_barcodes)
## filter
visium_brain <- filterGiotto(gobject = visium_brain,
expression_threshold = 1,
gene_det_in_min_cells = 50,
min_det_genes_per_cell = 1000,
expression_values = c('raw'),
verbose = T)
## normalize
visium_brain <- normalizeGiotto(gobject = visium_brain, scalefactor = 6000, verbose = T)
## add gene & cell statistics
visium_brain <- addStatistics(gobject = visium_brain)
## visualize
patPlot2D(gobject = visium_brain, show_image = T, point_alpha = 0.7,
ave_param = list(save_name = '2_d_spatial_locations'))
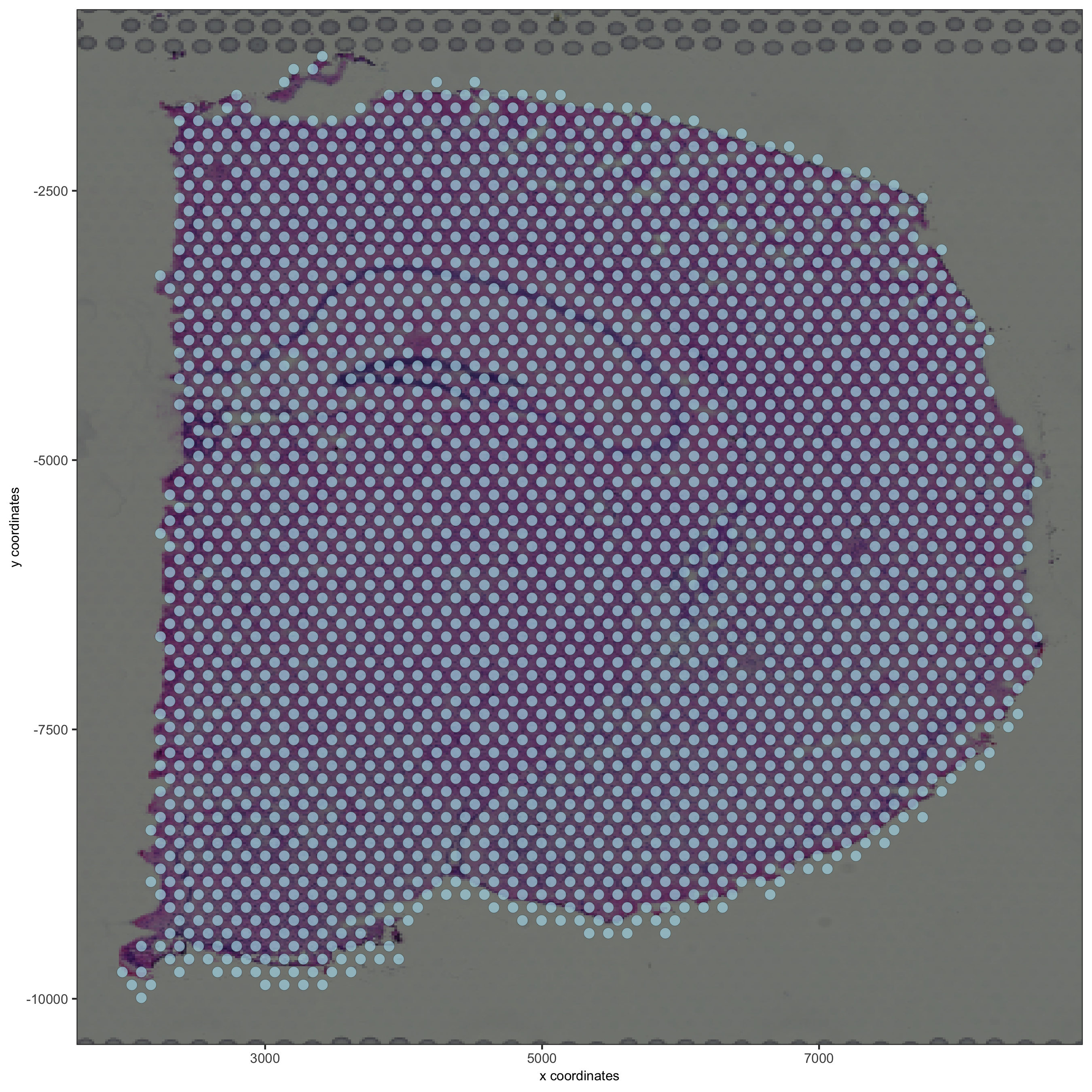
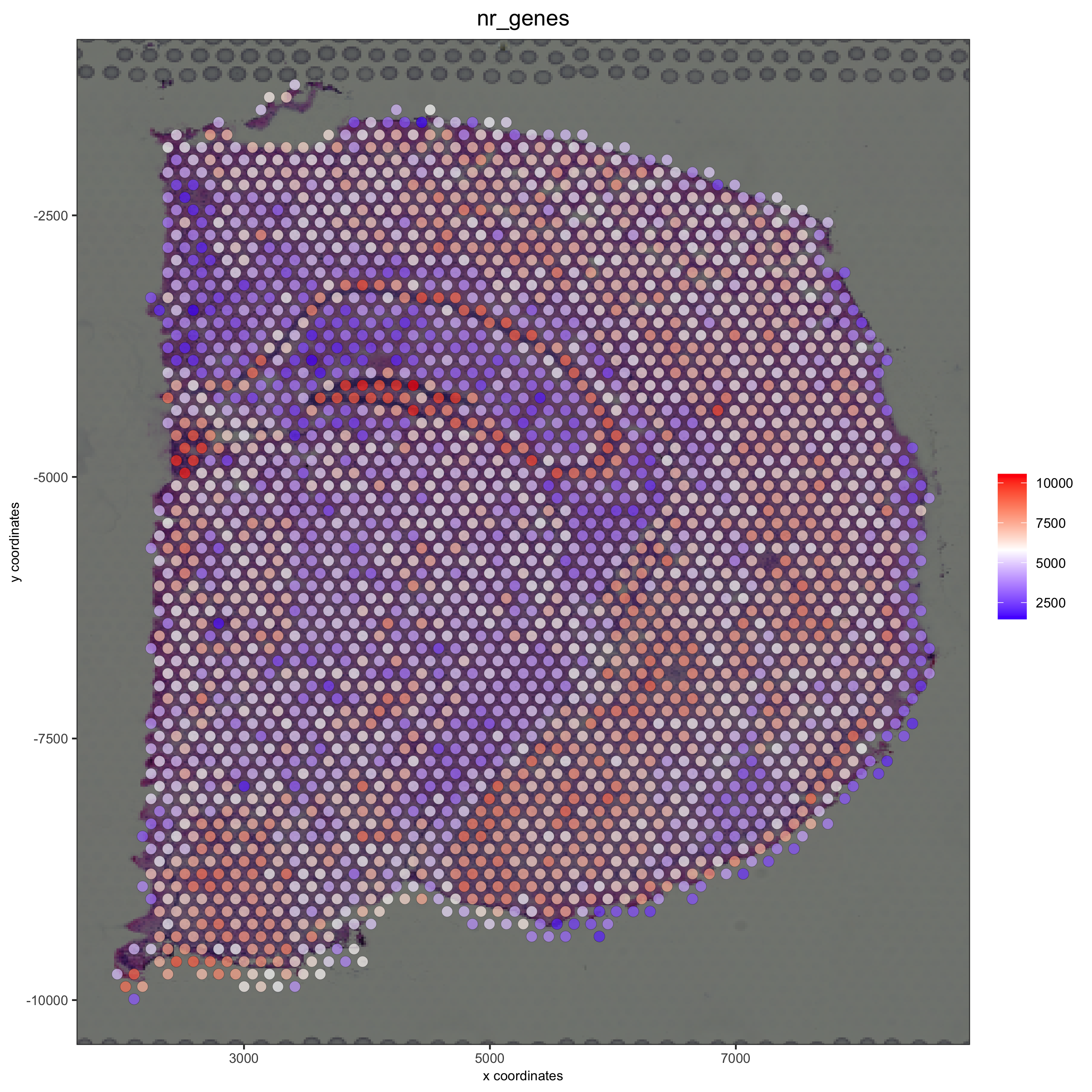
spatPlot2D(gobject = visium_brain, show_image = T, point_alpha = 0.7,
cell_color = 'nr_genes', color_as_factor = F,
save_param = list(save_name = '2_e_nr_genes'))
3. Dimension Reduction¶
## highly variable genes (HVG)
visium_brain <- calculateHVG(gobject = visium_brain,
save_param = list(save_name = '3_a_HVGplot'))
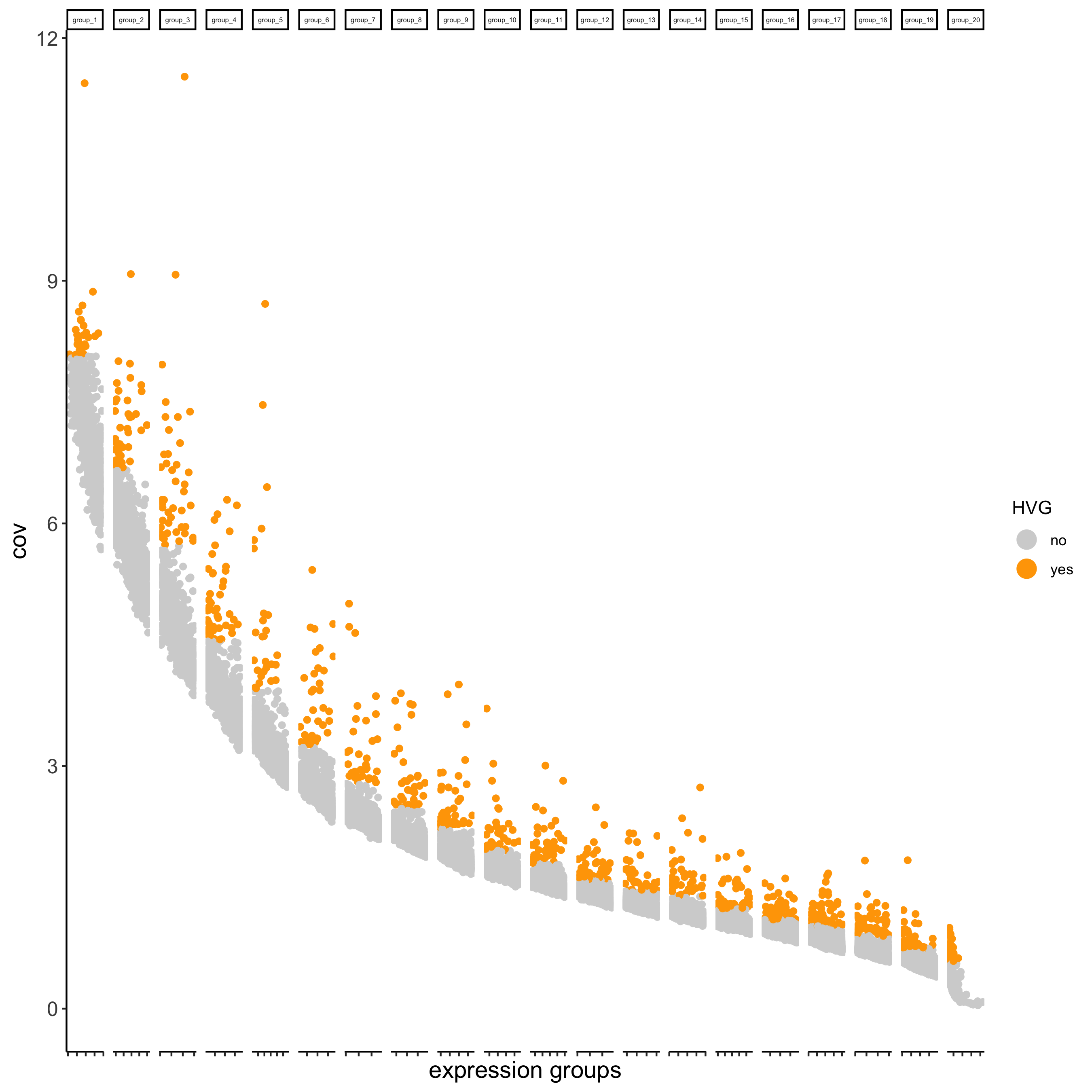
## run PCA on expression values (default)
gene_metadata = fDataDT(visium_brain)
featgenes = gene_metadata[hvg == 'yes' & perc_cells > 3 & mean_expr_det > 0.4]$gene_ID
visium_brain <- runPCA(gobject = visium_brain,
genes_to_use = featgenes,
scale_unit = F, center = T,
method="factominer")
screePlot(visium_brain, ncp = 30, save_param = list(save_name = '3_b_screeplot'))
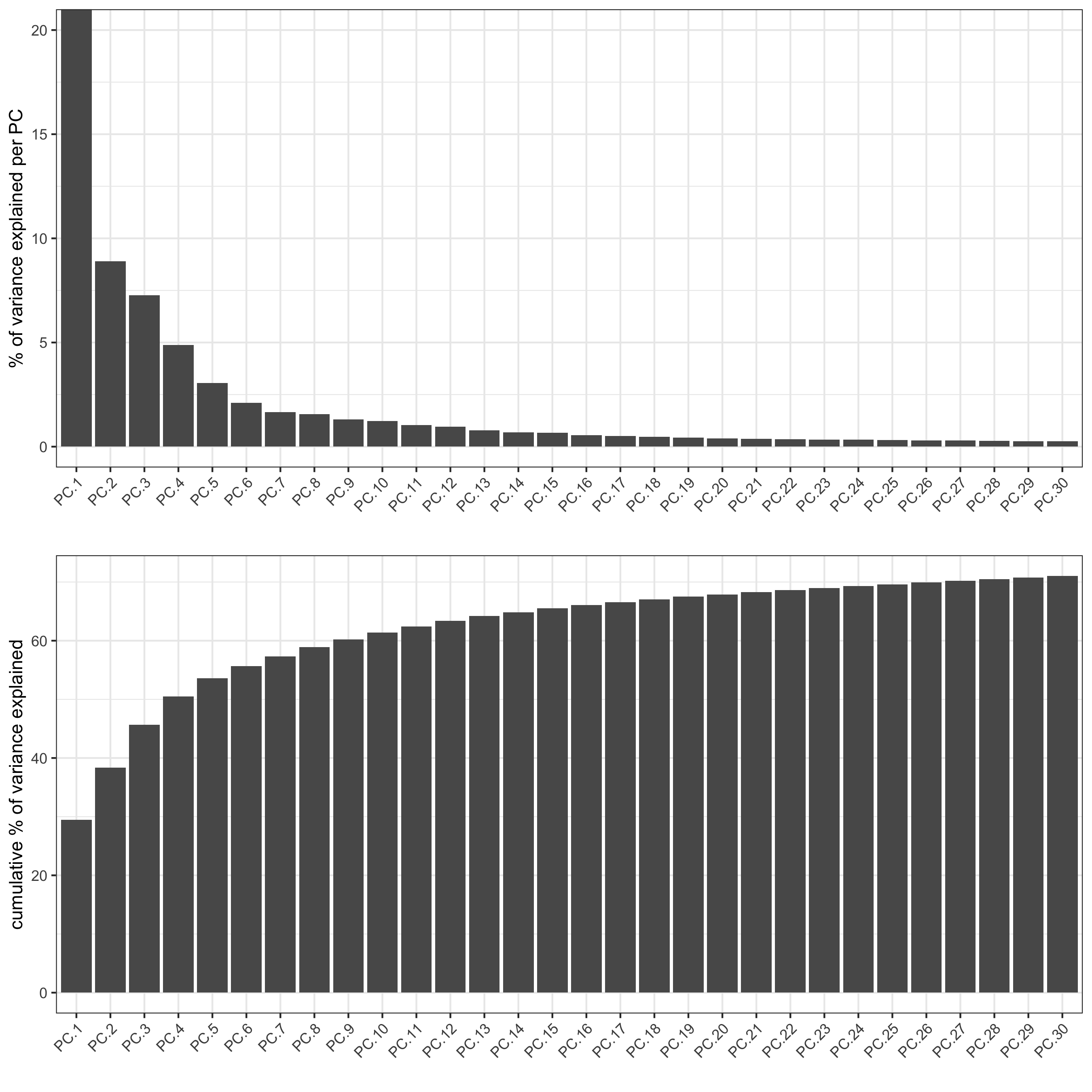
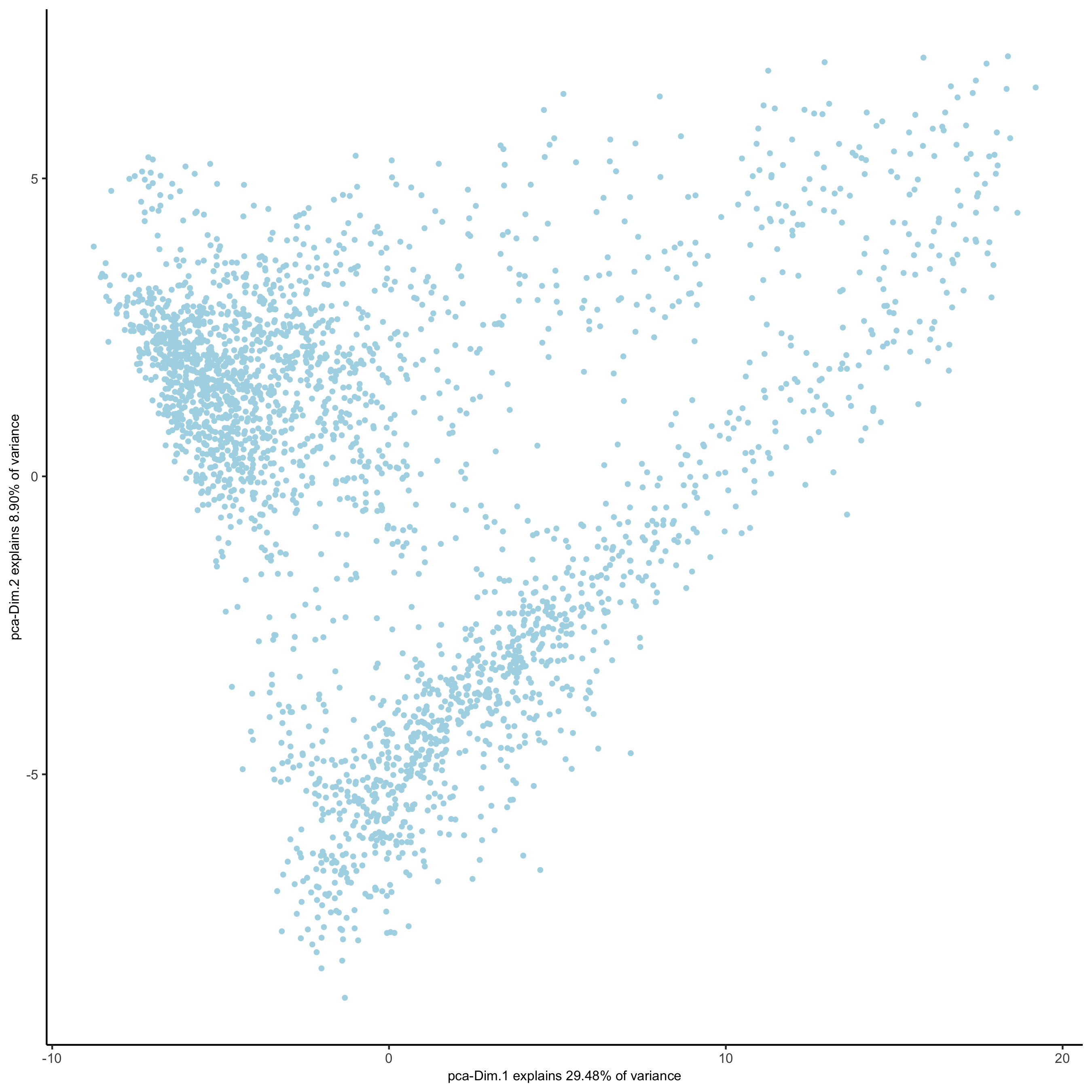
plotPCA(gobject = visium_brain,
save_param = list(save_name = '3_c_PCA_reduction'))
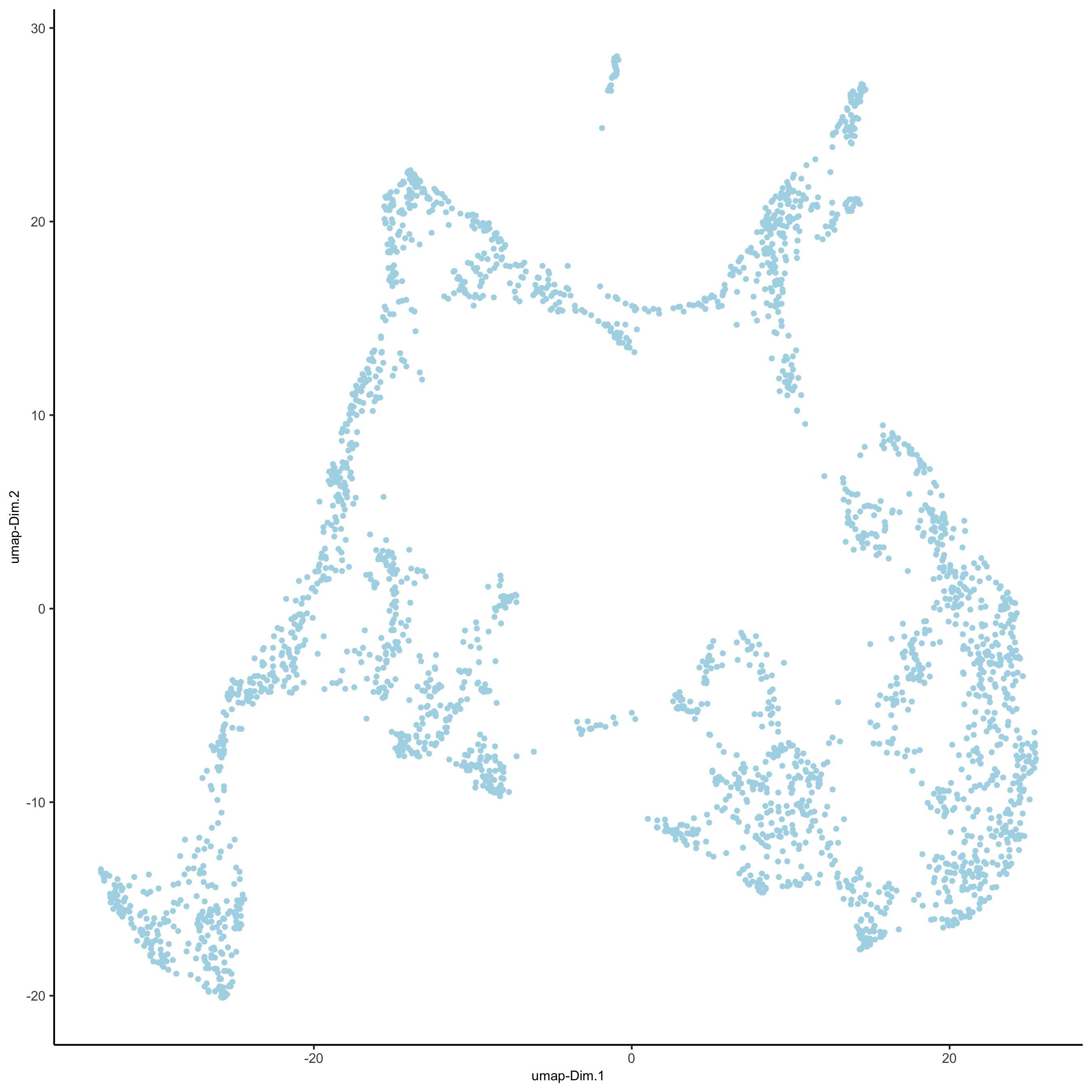
## run UMAP and tSNE on PCA space (default)
visium_brain <- runUMAP(visium_brain, dimensions_to_use = 1:10)
plotUMAP(gobject = visium_brain,
save_param = list(save_name = '3_d_UMAP_reduction'))
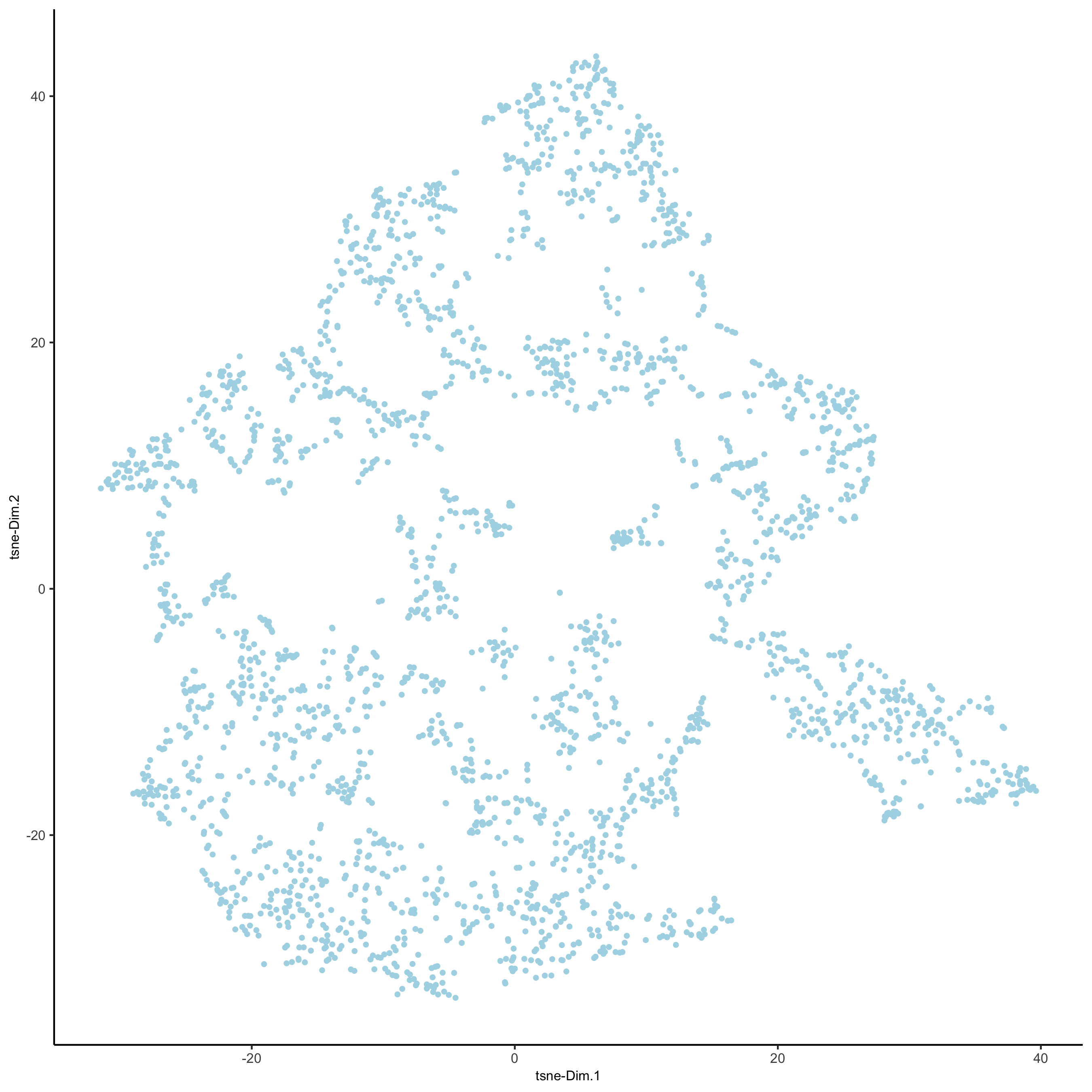
visium_brain <- runtSNE(visium_brain, dimensions_to_use = 1:10)
plotTSNE(gobject = visium_brain,
save_param = list(save_name = '3_e_tSNE_reduction'))
4. Clustering¶
## sNN network (default)
visium_brain <- createNearestNetwork(gobject = visium_brain, dimensions_to_use = 1:10, k = 15)
## Leiden clustering
visium_brain <- doLeidenCluster(gobject = visium_brain, resolution = 0.4, n_iterations = 1000)
plotUMAP(gobject = visium_brain,
cell_color = 'leiden_clus', show_NN_network = T, point_size = 2.5,
save_param = list(save_name = '4_a_UMAP_leiden'))
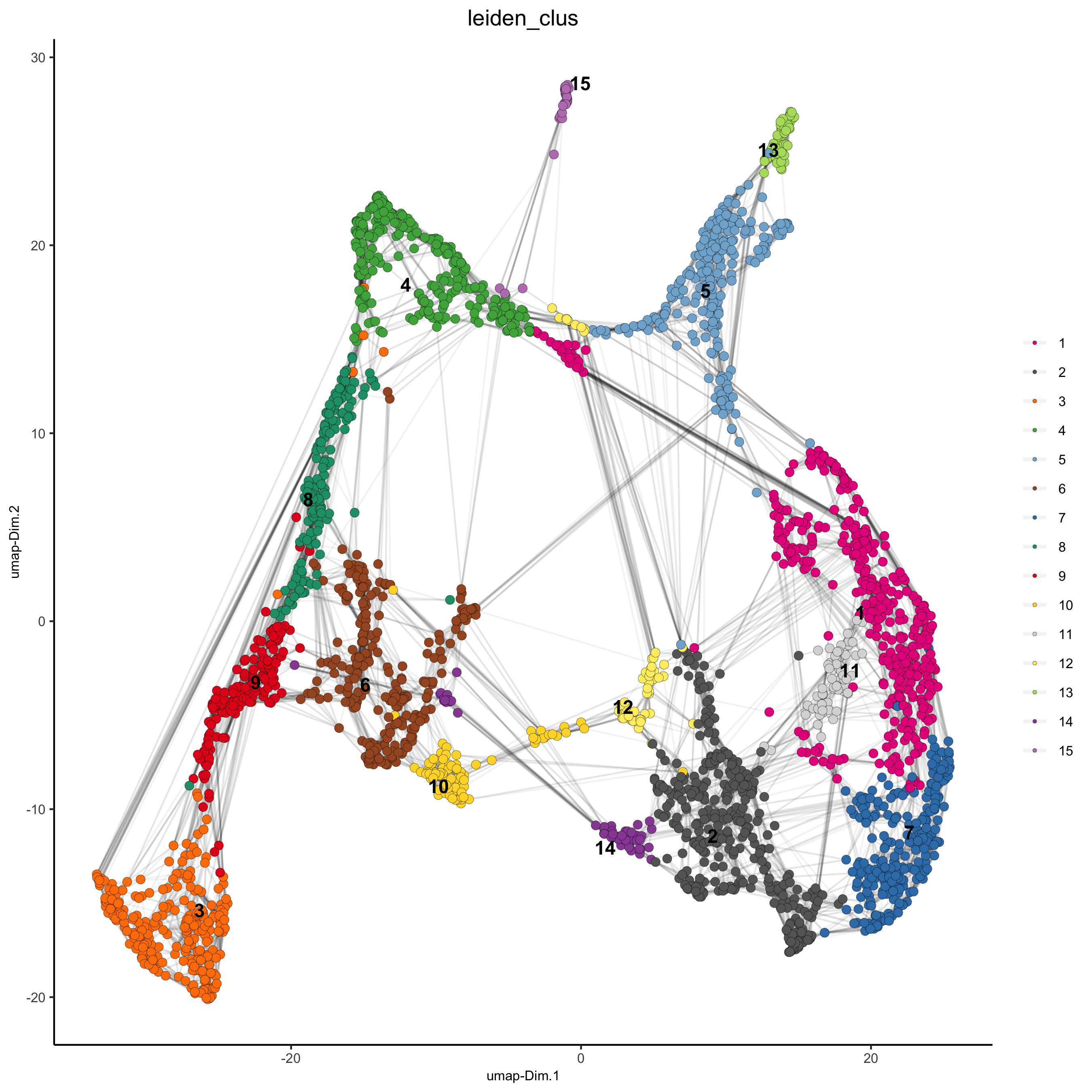
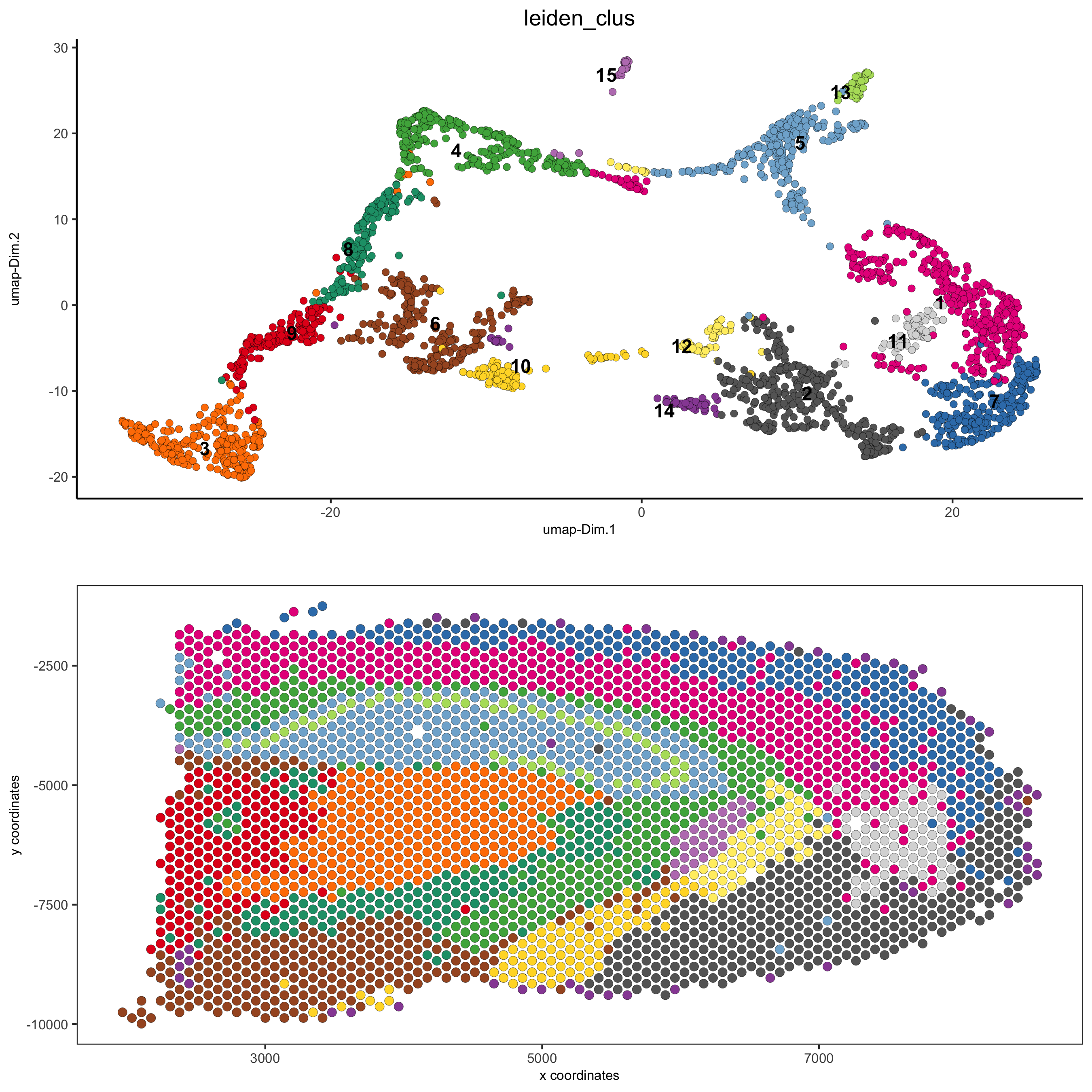
5. Whole Dataset¶
5.1 Expression and Spatial¶
# expression and spatial
spatDimPlot(gobject = visium_brain, cell_color = 'leiden_clus',
dim_point_size = 2, spat_point_size = 2.5,
save_param = list(save_name = '5_a_covis_leiden'))
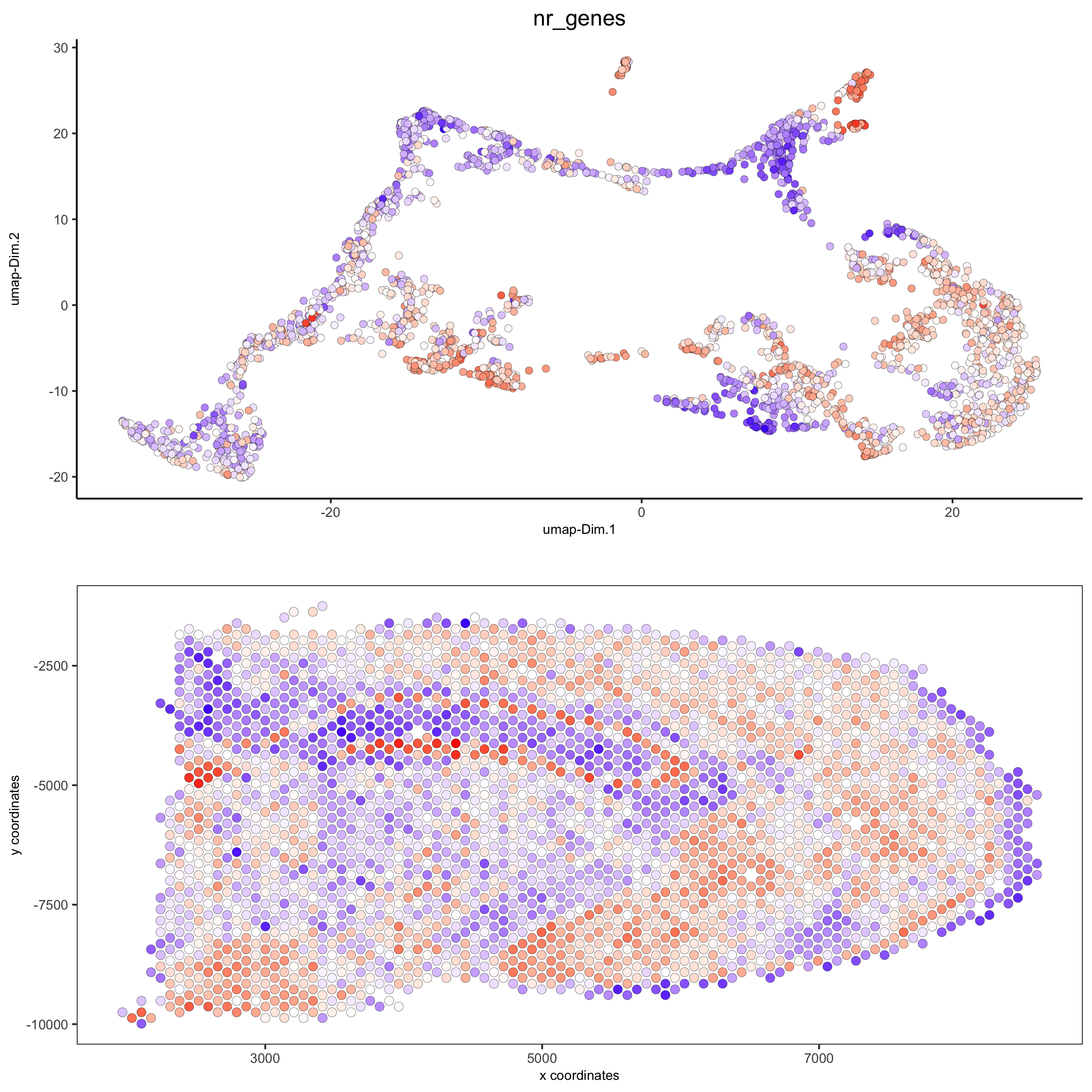
spatDimPlot(gobject = visium_brain, cell_color = 'nr_genes', color_as_factor = F,
dim_point_size = 2, spat_point_size = 2.5,
save_param = list(save_name = '5_b_nr_genes'))
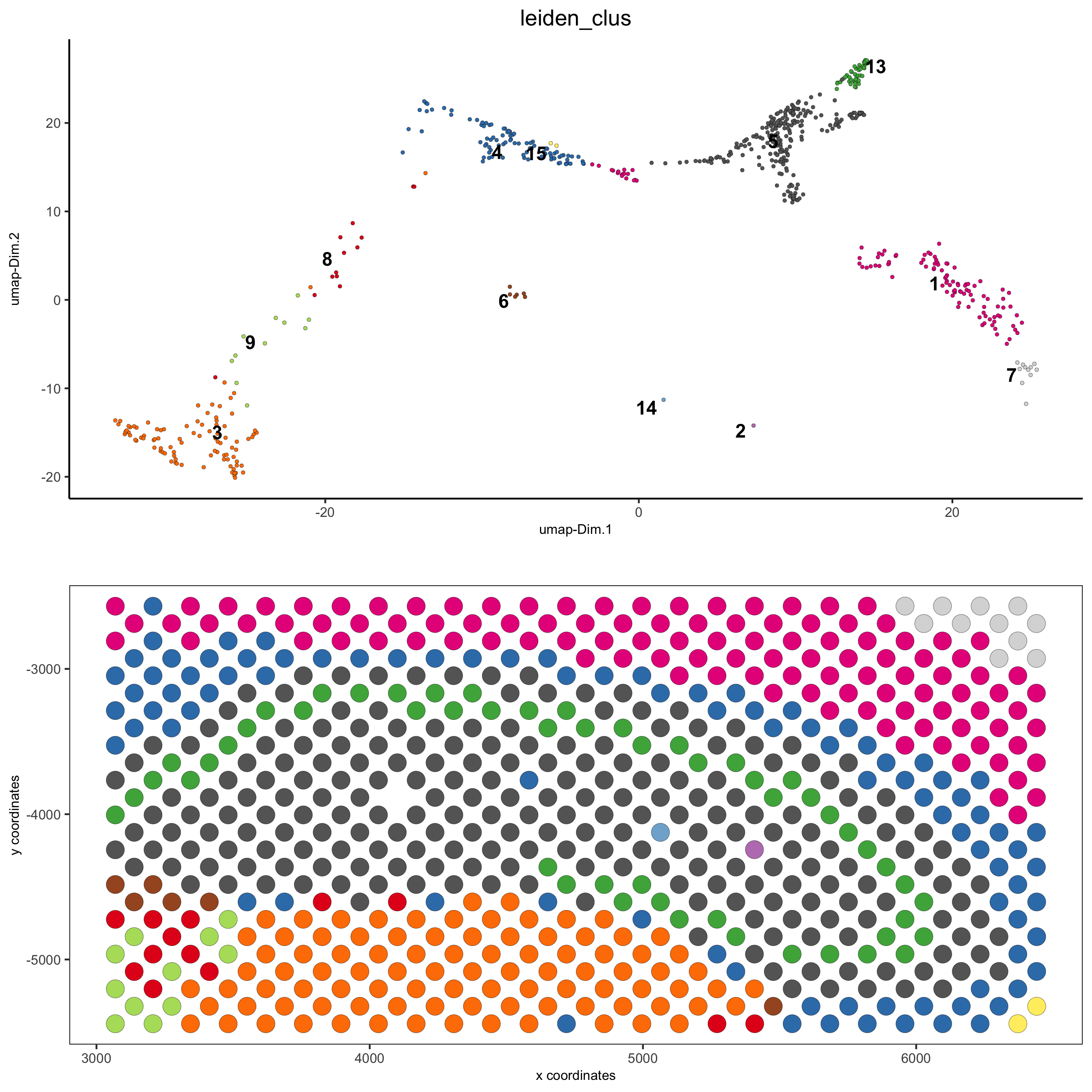
5.2 Subset Dataset on DG Region¶
DG_subset = subsetGiottoLocs(visium_brain,
x_max = 6500, x_min = 3000,
y_max = -2500, y_min = -5500,
return_gobject = TRUE)
spatDimPlot(gobject = DG_subset,
cell_color = 'leiden_clus', spat_point_size = 5,
save_param = list(save_name = '5_c_DEG_subset'))
6. Cell-Type Marker Gene Detection¶
6.1 Gini¶
gini_markers_subclusters = findMarkers_one_vs_all(gobject = visium_brain,
method = 'gini',
expression_values = 'normalized',
cluster_column = 'leiden_clus',
min_genes = 20,
min_expr_gini_score = 0.5,
min_det_gini_score = 0.5)
topgenes_gini = gini_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
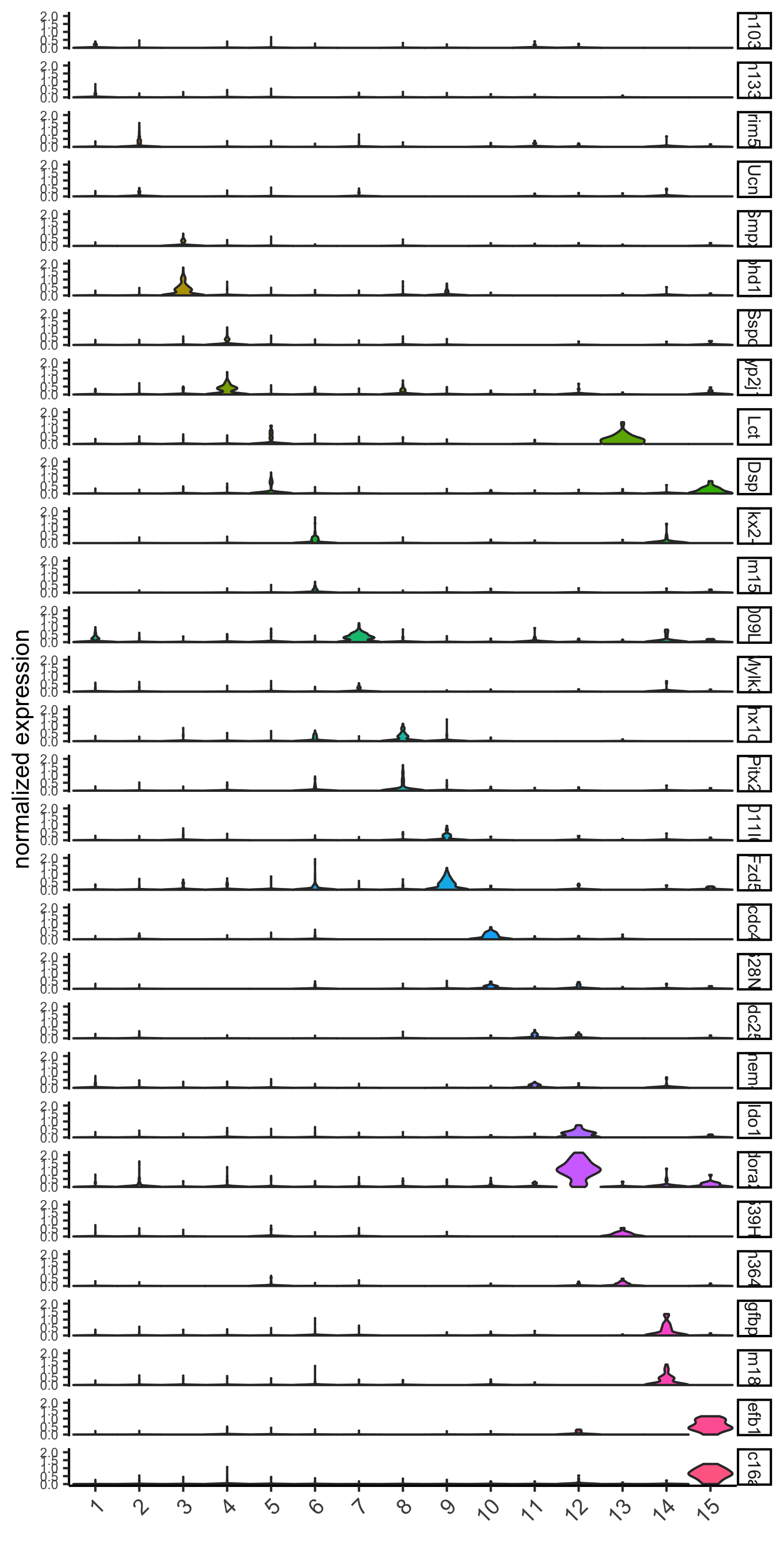
# violinplot
violinPlot(visium_brain, genes = unique(topgenes_gini), cluster_column = 'leiden_clus',
strip_text = 8, strip_position = 'right',
save_param = list(save_name = '6_a_violinplot_gini', base_width = 5, base_height = 10))
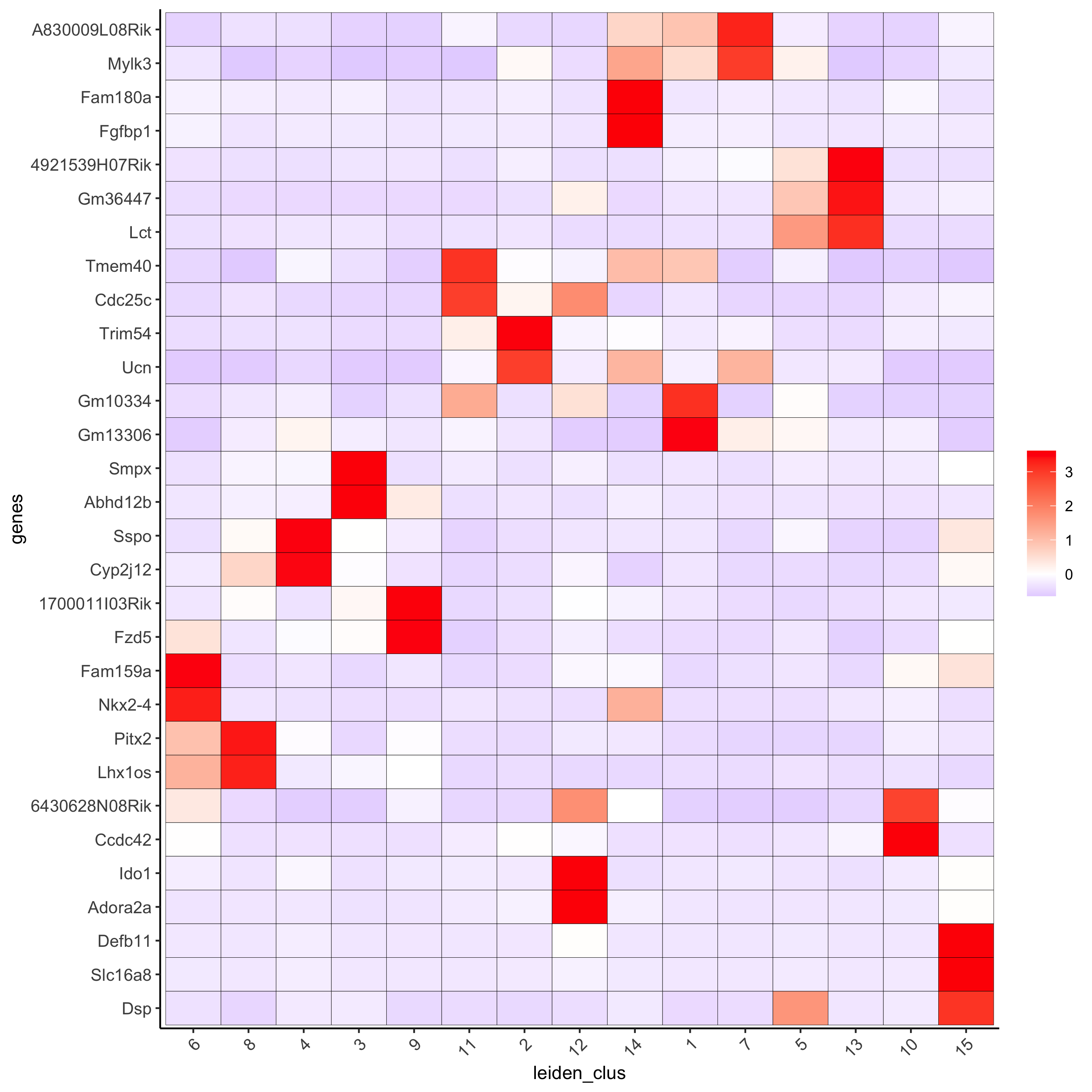
# cluster heatmap
plotMetaDataHeatmap(visium_brain, selected_genes = topgenes_gini,
metadata_cols = c('leiden_clus'),
x_text_size = 10, y_text_size = 10,
save_param = list(save_name = '6_b_metaheatmap_gini'))
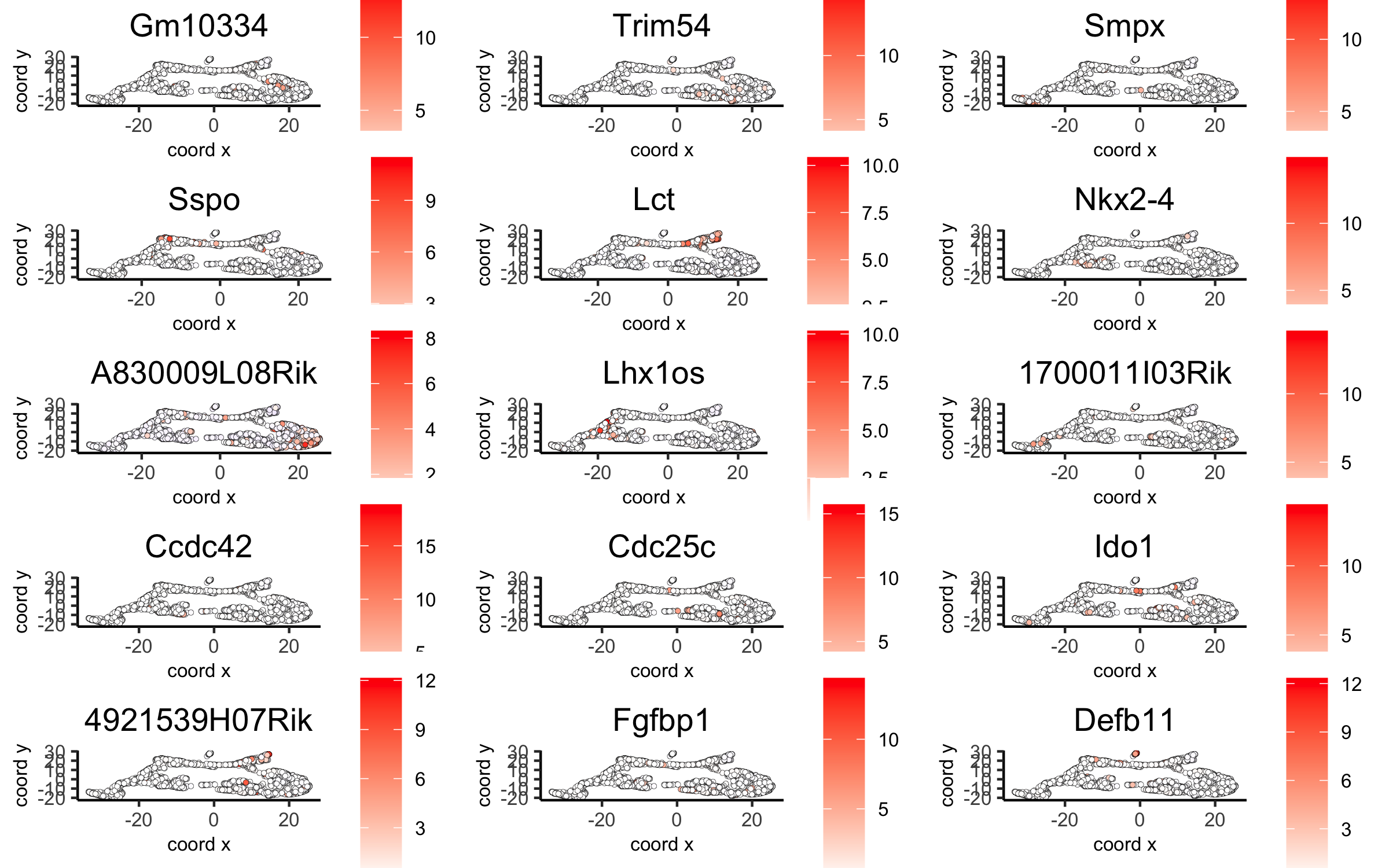
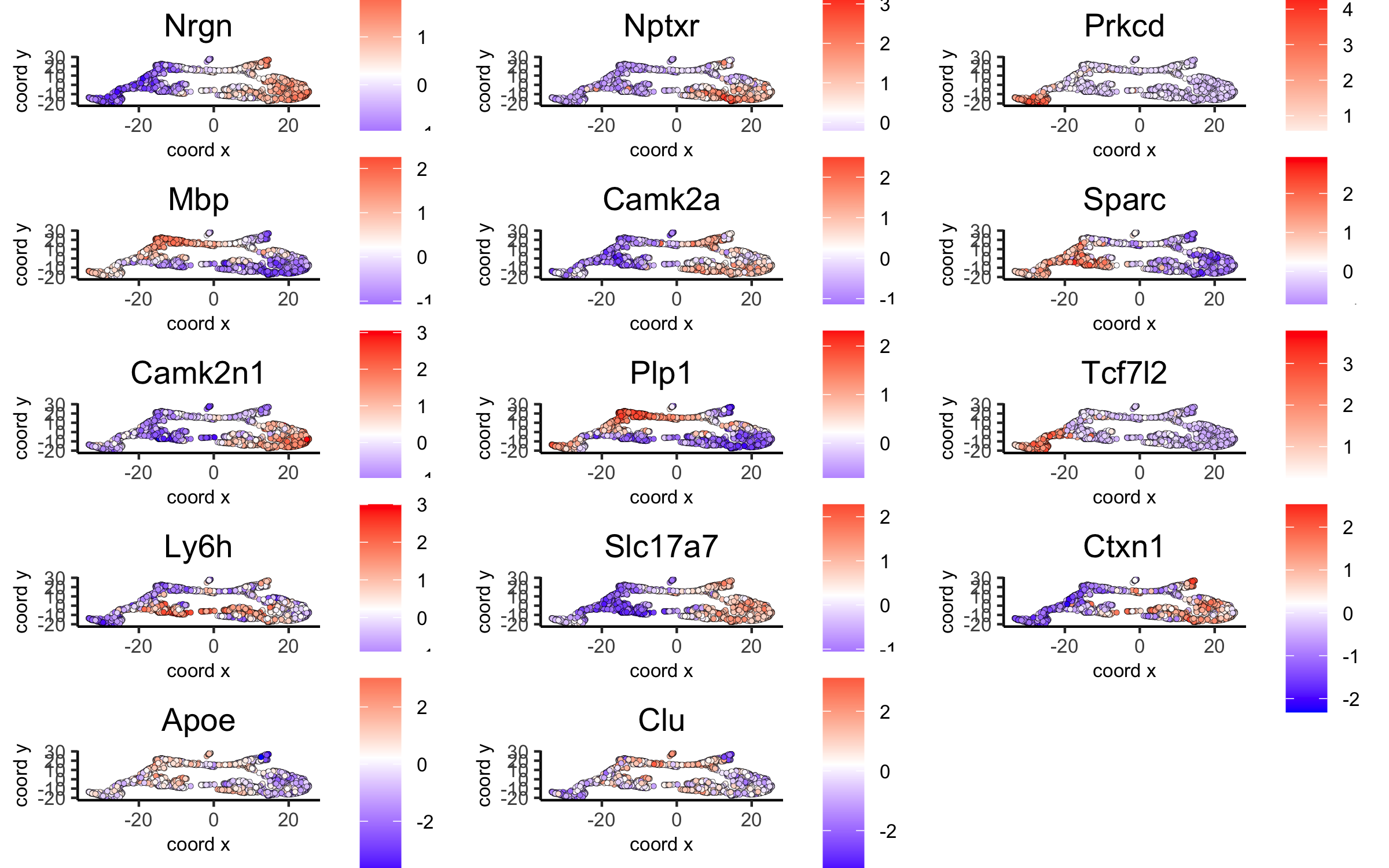
# umap plots
dimGenePlot2D(visium_brain, expression_values = 'scaled',
genes = gini_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,
cow_n_col = 3, point_size = 1,
save_param = list(save_name = '6_c_gini_umap', base_width = 8, base_height = 5))
6.2 Scran¶
scran_markers_subclusters = findMarkers_one_vs_all(gobject = visium_brain,
method = 'scran',
expression_values = 'normalized',
cluster_column = 'leiden_clus')
topgenes_scran = scran_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
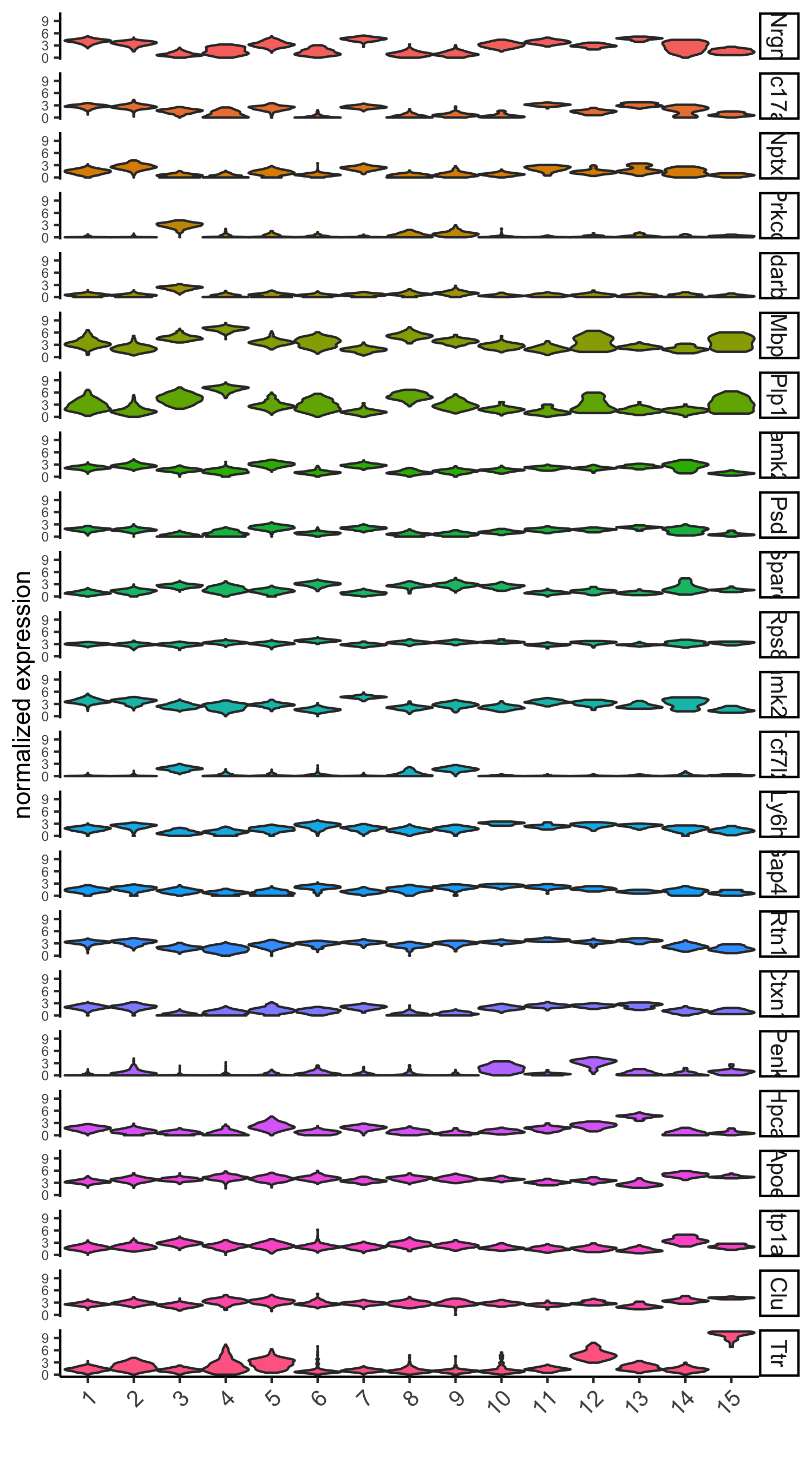
# violinplot
violinPlot(visium_brain, genes = unique(topgenes_scran), cluster_column = 'leiden_clus',
strip_text = 10, strip_position = 'right',
save_param = list(save_name = '6_d_violinplot_scran', base_width = 5))
# cluster heatmap
plotMetaDataHeatmap(visium_brain, selected_genes = topgenes_scran,
metadata_cols = c('leiden_clus'),
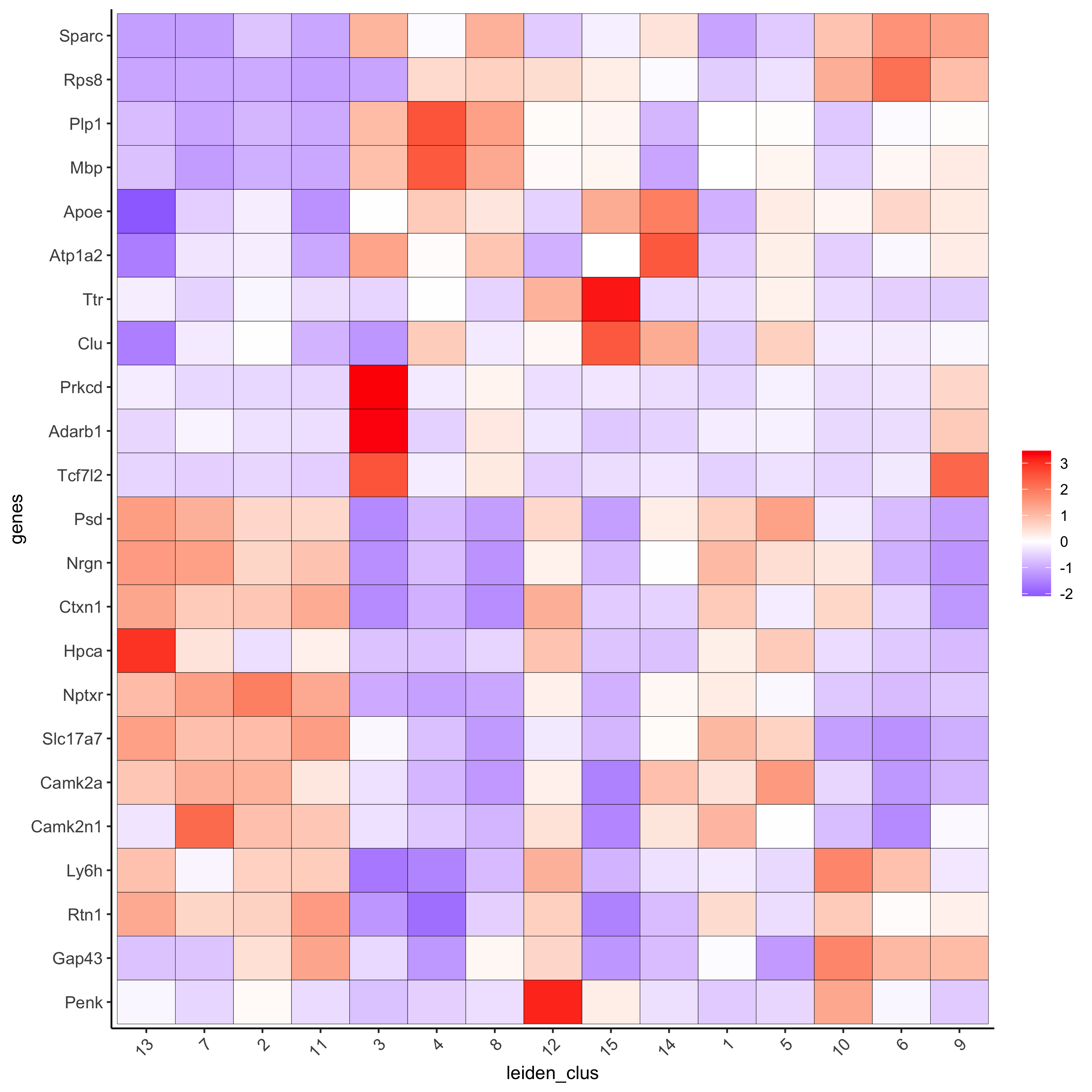
# umap plots
dimGenePlot(visium_brain, expression_values = 'scaled',
genes = scran_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,
cow_n_col = 3, point_size = 1,
save_param = list(save_name = '6_f_scran_umap', base_width = 8, base_height = 5))
7. Cell-Type Annotation¶
Visium spatial transcriptomics does not provide single-cell resolution, making cell type annotation a harder problem. Giotto provides 3 ways to calculate enrichment of specific cell-type signature gene list:
PAGE
RANK
Hypergeometric Test
Known markers for different mouse brain cell types: Zeisel, A. et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999-1014.e22 (2018) . Cell type signatures are combination of all marker genes identified in Zeisel et al.
7.1 PAGE Enrichment¶
# 1.1 create binary matrix of cell signature genes
# small example #
gran_markers = c("Nr3c2", "Gabra5", "Tubgcp2", "Ahcyl2",
"Islr2", "Rasl10a", "Tmem114", "Bhlhe22",
"Ntf3", "C1ql2")
oligo_markers = c("Efhd1", "H2-Ab1", "Enpp6", "Ninj2",
"Bmp4", "Tnr", "Hapln2", "Neu4",
"Wfdc18", "Ccp110")
di_mesench_markers = c("Cartpt", "Scn1a", "Lypd6b", "Drd5",
"Gpr88", "Plcxd2", "Cpne7", "Pou4f1",
"Ctxn2", "Wnt4")
signature_matrix = makeSignMatrixPAGE(sign_names = c('Granule_neurons',
'Oligo_dendrocytes',
'di_mesenchephalon'),
sign_list = list(gran_markers,
oligo_markers,
di_mesench_markers))
# 1.2 [shortcut] fully pre-prepared matrix for all cell types
sign_matrix_path = system.file("extdata", "sig_matrix.txt", package = 'Giotto')
brain_sc_markers = data.table::fread(sign_matrix_path)
sig_matrix = as.matrix(brain_sc_markers[,-1]); rownames(sig_matrix) = brain_sc_markers$Event
# 1.3 enrichment test with PAGE
# runSpatialEnrich() can also be used as a wrapper for all currently provided enrichment options
visium_brain = runPAGEEnrich(gobject = visium_brain, sign_matrix = sig_matrix)
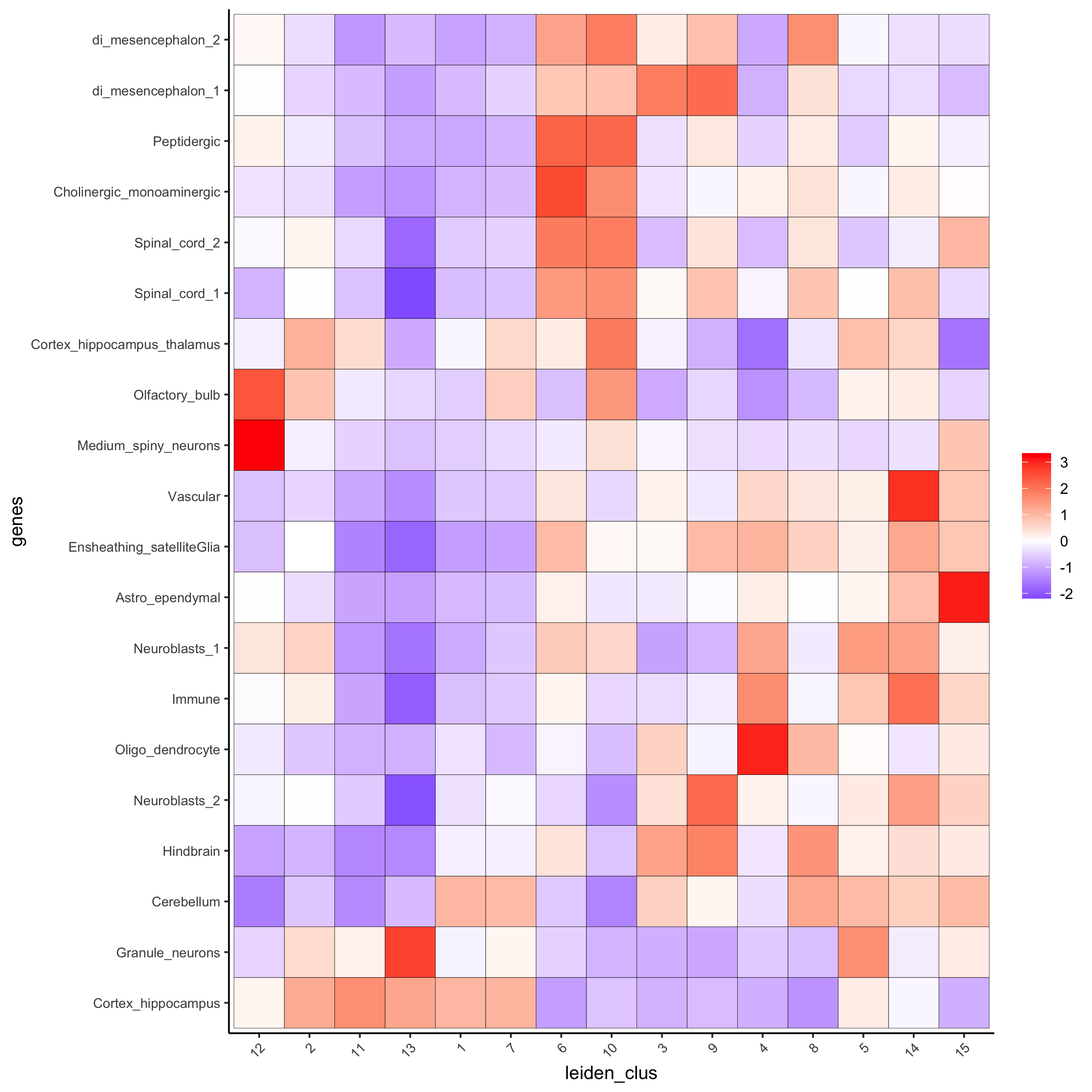
# 1.4 heatmap of enrichment versus annotation (e.g. clustering result)
cell_types = colnames(sig_matrix)
plotMetaDataCellsHeatmap(gobject = visium_brain,
metadata_cols = 'leiden_clus',
value_cols = cell_types,
spat_enr_names = 'PAGE',
x_text_size = 8,
y_text_size = 8,
save_param = list(save_name="7_a_metaheatmap"))
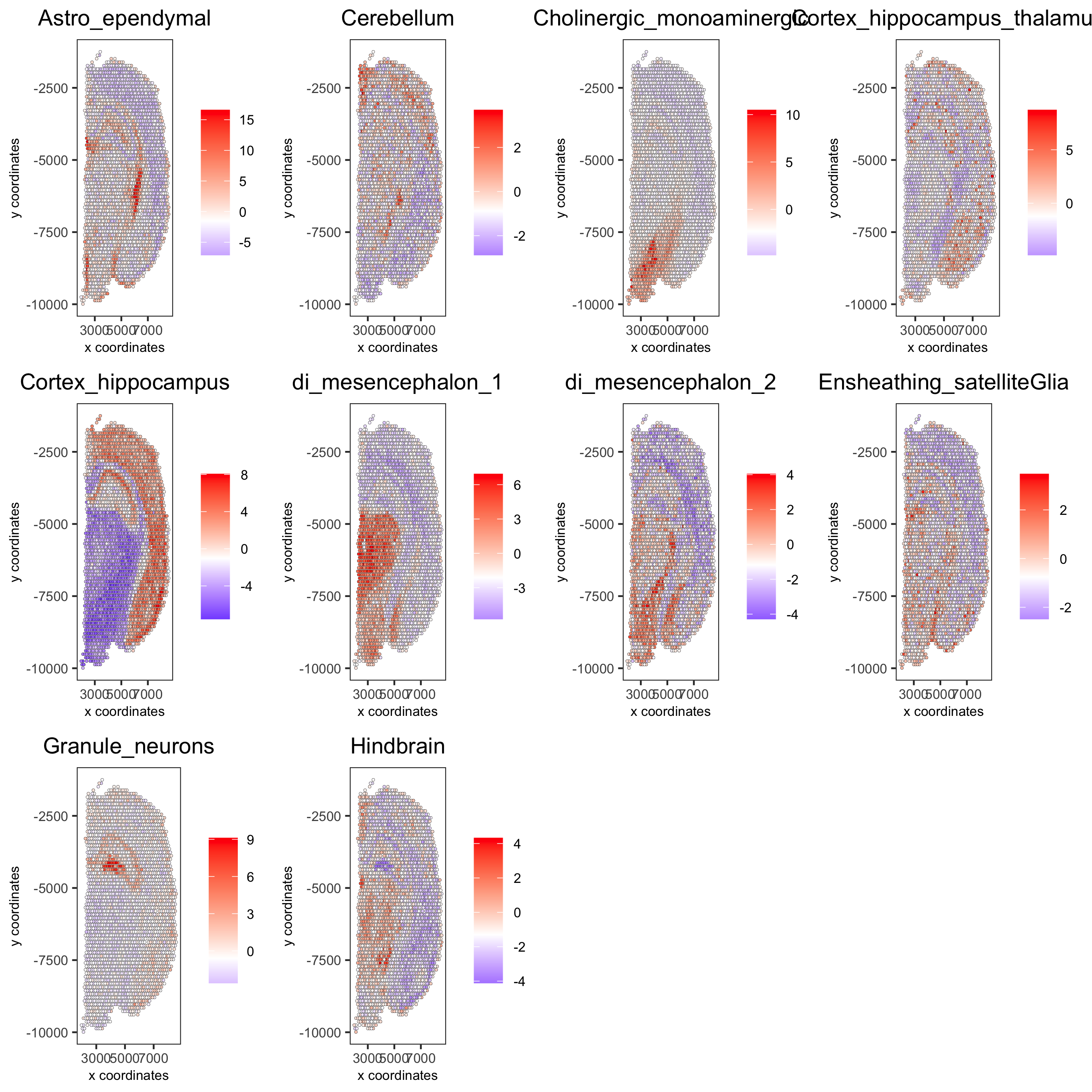
# 1.5 visualizations
cell_types_subset = colnames(sig_matrix)[1:10]
spatCellPlot(gobject = visium_brain,
spat_enr_names = 'PAGE',
cell_annotation_values = cell_types_subset,
cow_n_col = 4,coord_fix_ratio = NULL, point_size = 0.75,
save_param = list(save_name="7_b_spatcellplot_1"))
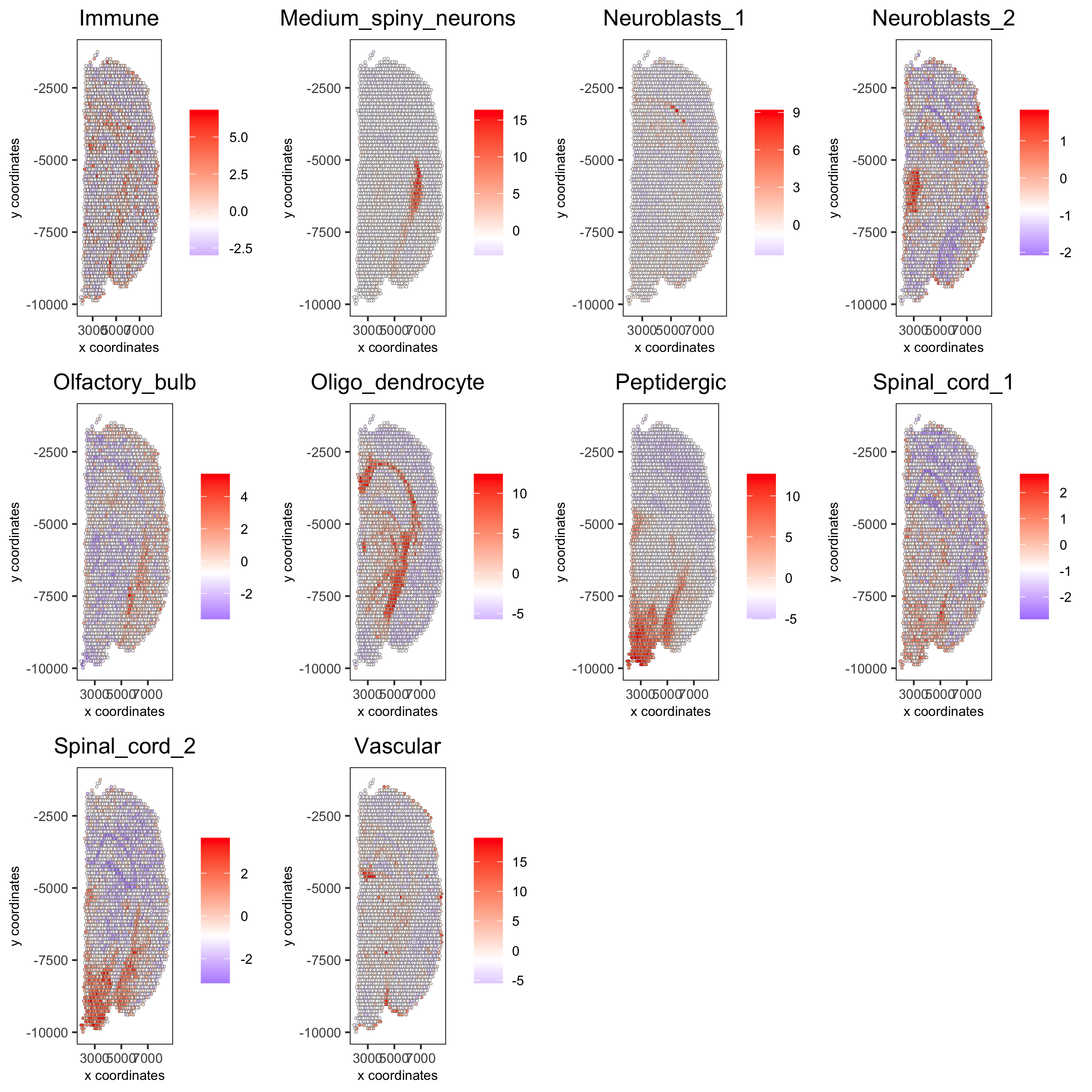
cell_types_subset = colnames(sig_matrix)[11:20]
spatCellPlot(gobject = visium_brain, spat_enr_names = 'PAGE',
cell_annotation_values = cell_types_subset, cow_n_col = 4,
coord_fix_ratio = NULL, point_size = 0.75,
save_param = list(save_name="7_c_spatcellplot_2"))
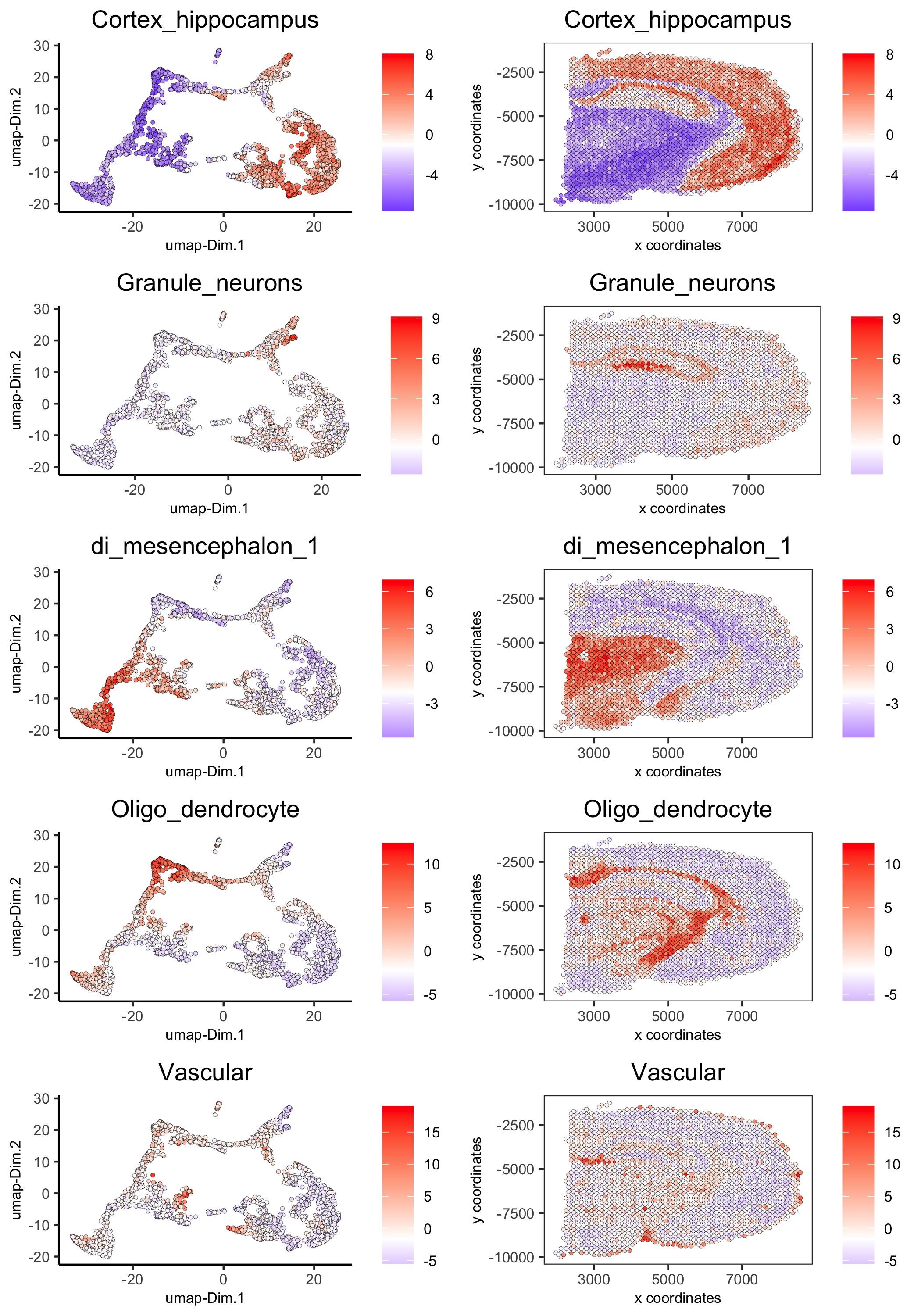
spatDimCellPlot(gobject = visium_brain,
spat_enr_names = 'PAGE',
cell_annotation_values = c('Cortex_hippocampus', 'Granule_neurons',
'di_mesencephalon_1', 'Oligo_dendrocyte','Vascular'),
cow_n_col = 1, spat_point_size = 1,
plot_alignment = 'horizontal',
save_param = list(save_name="7_d_spatDimCellPlot", base_width=7, base_height=10))
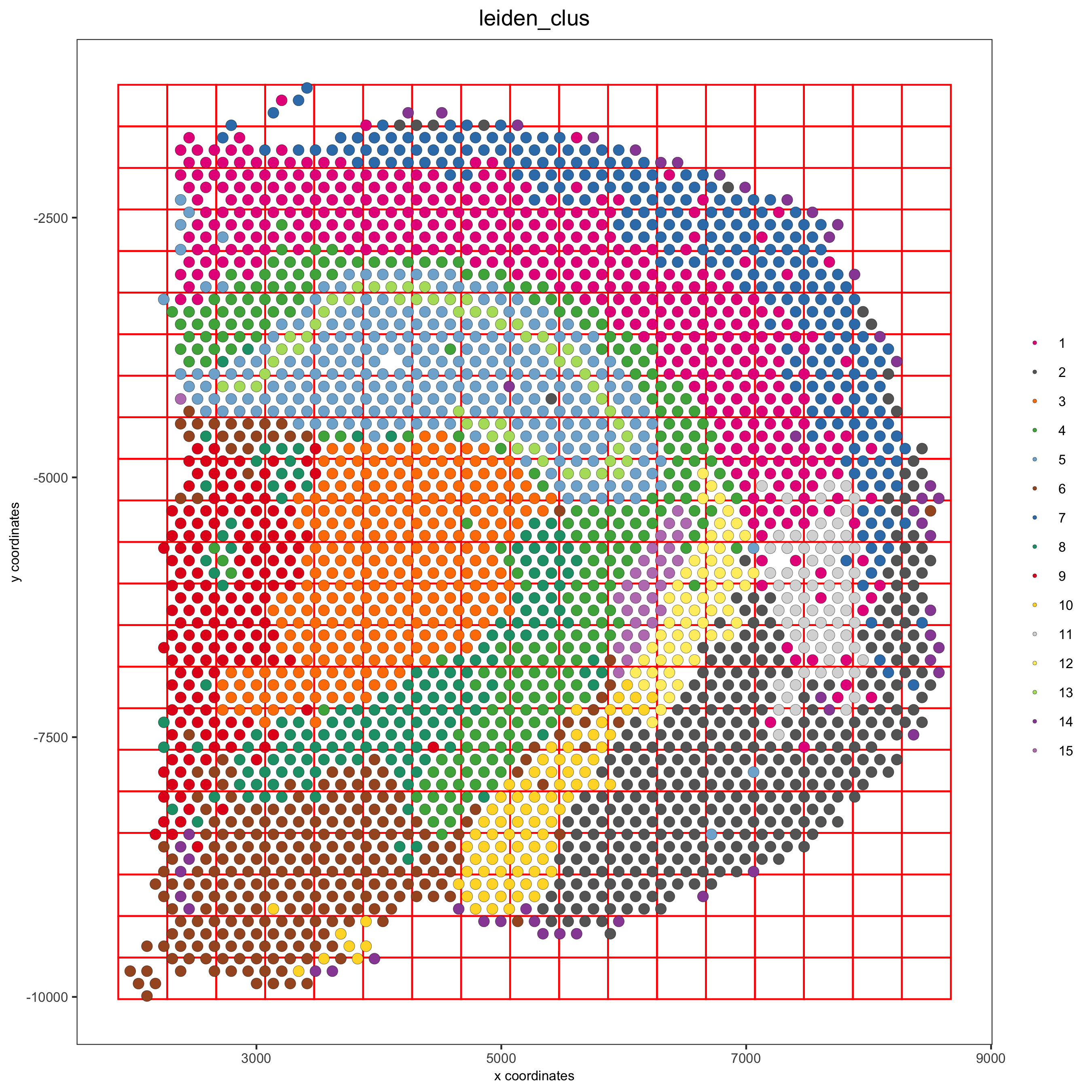
8. Spatial Grid¶
visium_brain <- createSpatialGrid(gobject = visium_brain,
sdimx_stepsize = 400,
sdimy_stepsize = 400,
minimum_padding = 0)
spatPlot(visium_brain, cell_color = 'leiden_clus', show_grid = T,
grid_color = 'red', spatial_grid_name = 'spatial_grid',
save_param = list(save_name = '8_grid'))
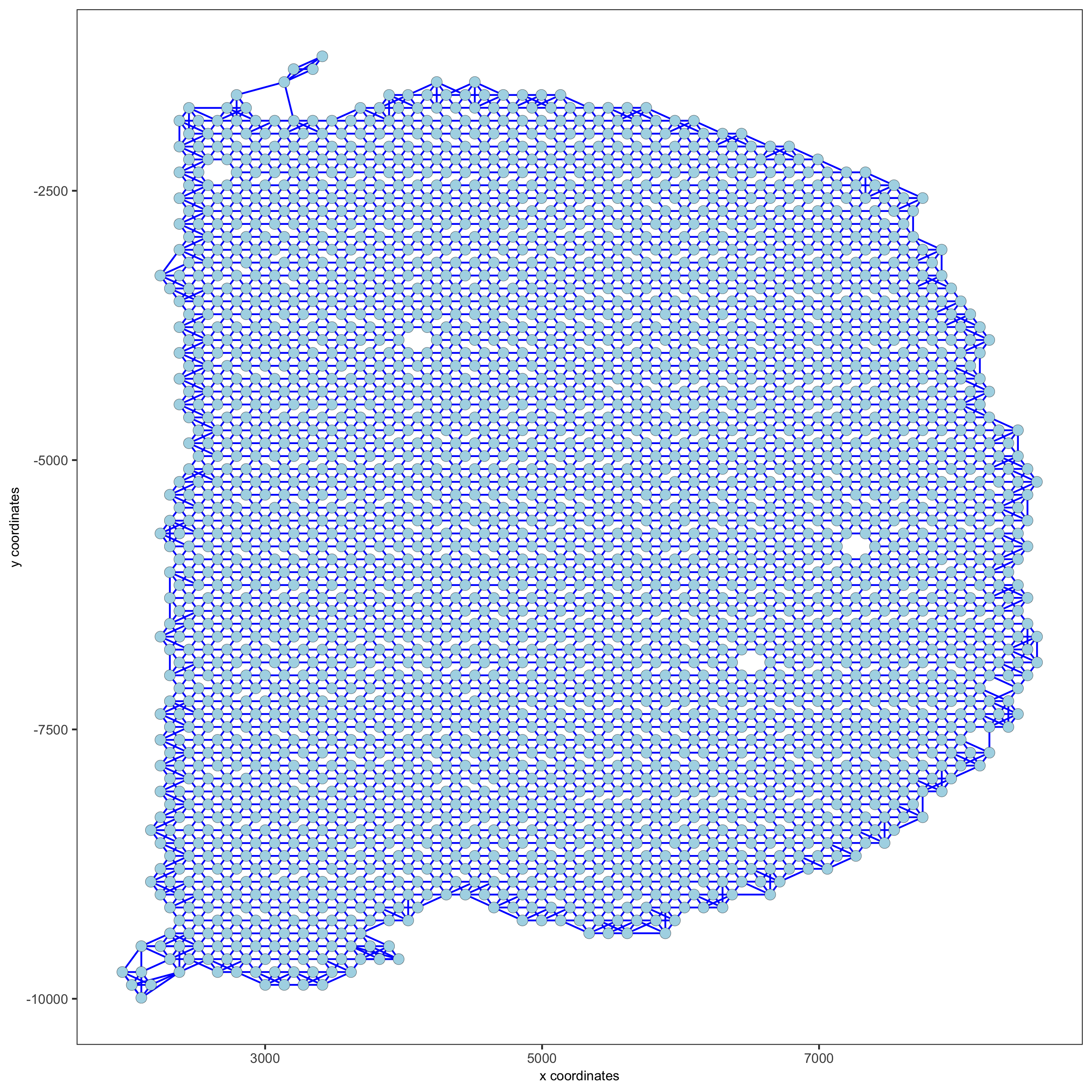
9. Spatial Network¶
visium_brain <- createSpatialNetwork(gobject = visium_brain,
method = 'kNN', k = 5,
maximum_distance_knn = 400,
name = 'spatial_network')
showNetworks(visium_brain)
spatPlot(gobject = visium_brain, show_network = T,
network_color = 'blue', spatial_network_name = 'spatial_network',
save_param = list(save_name = '9_a_knn_network'))
10. Spatial Genes and Patterns¶
10.1 Spatial Genes¶
## kmeans binarization
kmtest = binSpect(visium_brain, calc_hub = T, hub_min_int = 5,
spatial_network_name = 'spatial_network')
spatGenePlot(visium_brain, expression_values = 'scaled',
genes = kmtest$genes[1:6], cow_n_col = 2, point_size = 1.5,
save_param = list(save_name = '10_a_spatial_genes_km'))
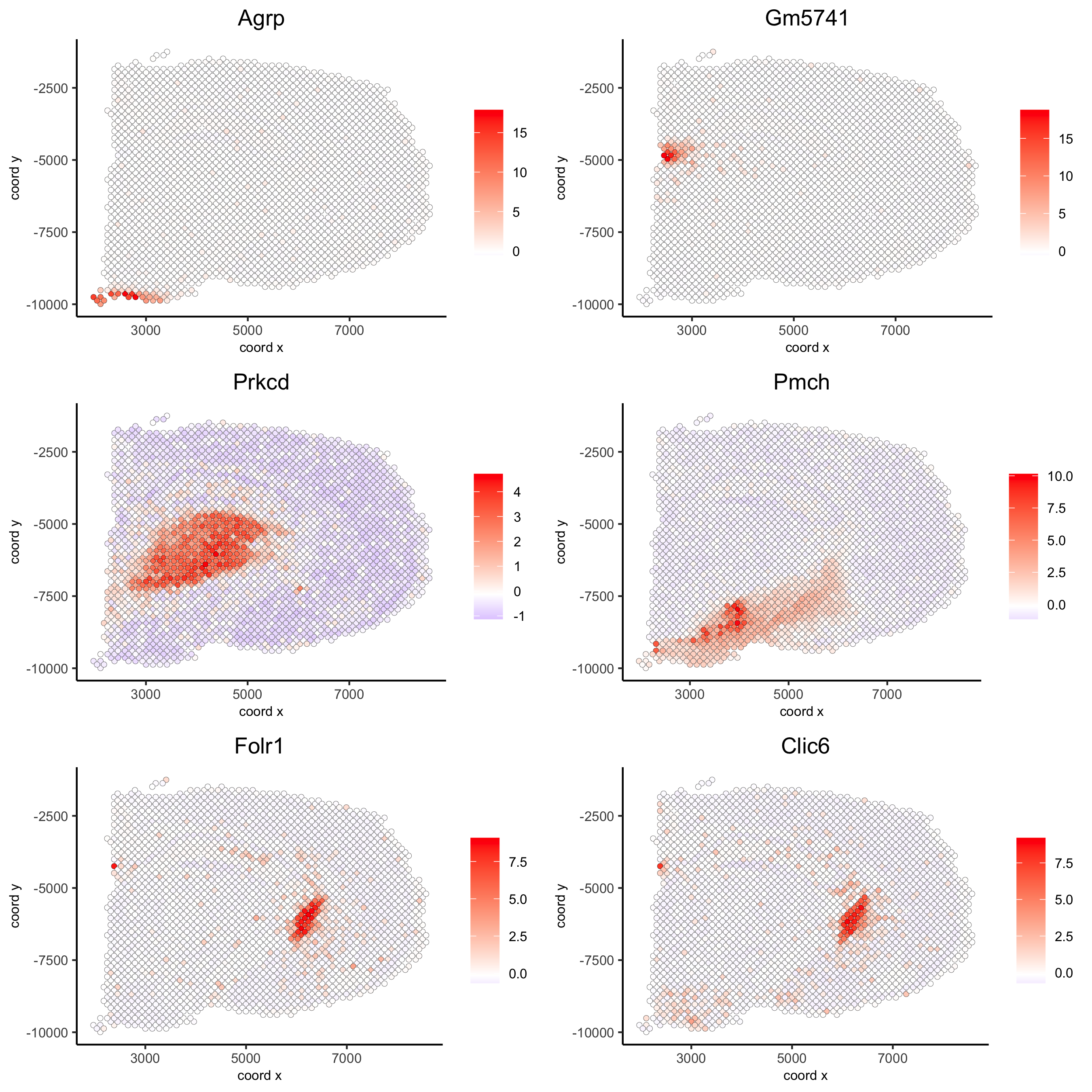
## rank binarization
ranktest = binSpect(visium_brain, bin_method = 'rank',
calc_hub = T, hub_min_int = 5,
spatial_network_name = 'spatial_network')
spatGenePlot(visium_brain, expression_values = 'scaled',
genes = ranktest$genes[1:6], cow_n_col = 2, point_size = 1.5,
save_param = list(save_name = '10_b_spatial_genes_rank'))
10.2 Spatial Patterns¶
# cluster the top 1500 spatial genes into 20 clusters
ext_spatial_genes = ranktest[1:1500,]$gene
# here we use existing detectSpatialCorGenes function to calculate pairwise distances between genes (but set network_smoothing=0 to use default clustering)
spat_cor_netw_DT = detectSpatialCorGenes(visium_brain,
method = 'network',
spatial_network_name = 'spatial_network',
subset_genes = ext_spatial_genes)
# cluster spatial genes
spat_cor_netw_DT = clusterSpatialCorGenes(spat_cor_netw_DT, name = 'spat_netw_clus', k = 20)
# visualize clusters
heatmSpatialCorGenes(visium_brain,
spatCorObject = spat_cor_netw_DT,
use_clus_name = 'spat_netw_clus',
heatmap_legend_param = list(title = NULL),
save_param = list(save_name="10_c_heatmap",
base_height = 6, base_width = 8, units = 'cm'))
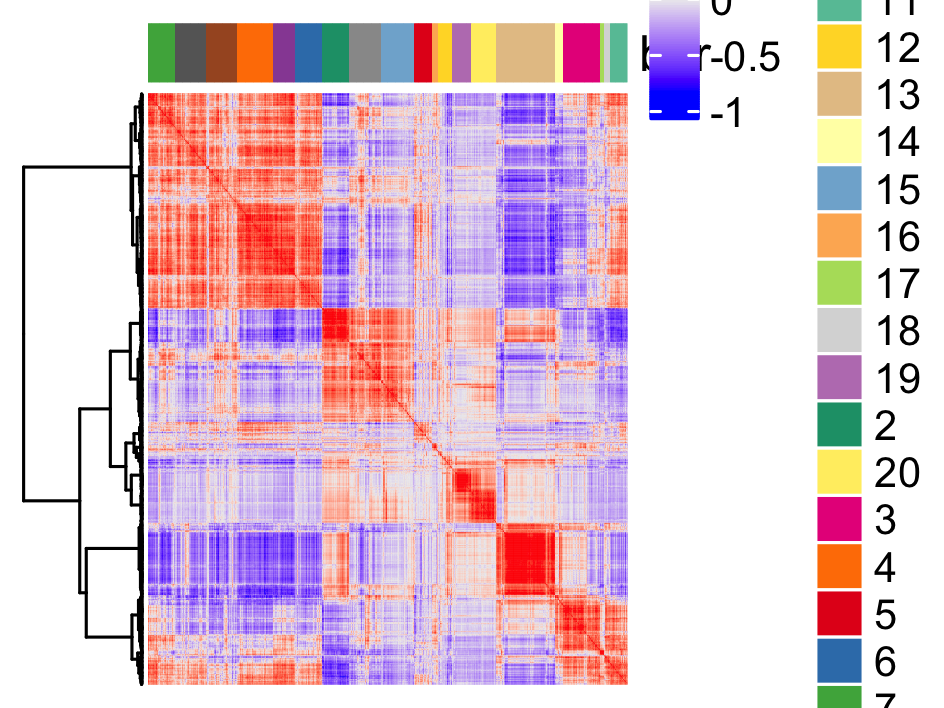
table(spat_cor_netw_DT$cor_clusters$spat_netw_clus)
coexpr_dt = data.table::data.table(genes = names(spat_cor_netw_DT$cor_clusters$spat_netw_clus),
cluster = spat_cor_netw_DT$cor_clusters$spat_netw_clus)
data.table::setorder(coexpr_dt, cluster)
top30_coexpr_dt = coexpr_dt[, head(.SD, 30) , by = cluster]
# do HMRF with different betas on 500 spatial genes
my_spatial_genes <- top30_coexpr_dt$genes
hmrf_folder = paste0(results_folder,'/','11_HMRF/')
if(!file.exists(hmrf_folder)) dir.create(hmrf_folder, recursive = T)
HMRF_spatial_genes = doHMRF(gobject = visium_brain,
expression_values = 'scaled',
spatial_genes = my_spatial_genes, k = 20,
spatial_network_name="spatial_network",
betas = c(0, 10, 5),
output_folder = paste0(hmrf_folder, '/', 'Spatial_genes/SG_topgenes_k20_scaled'))
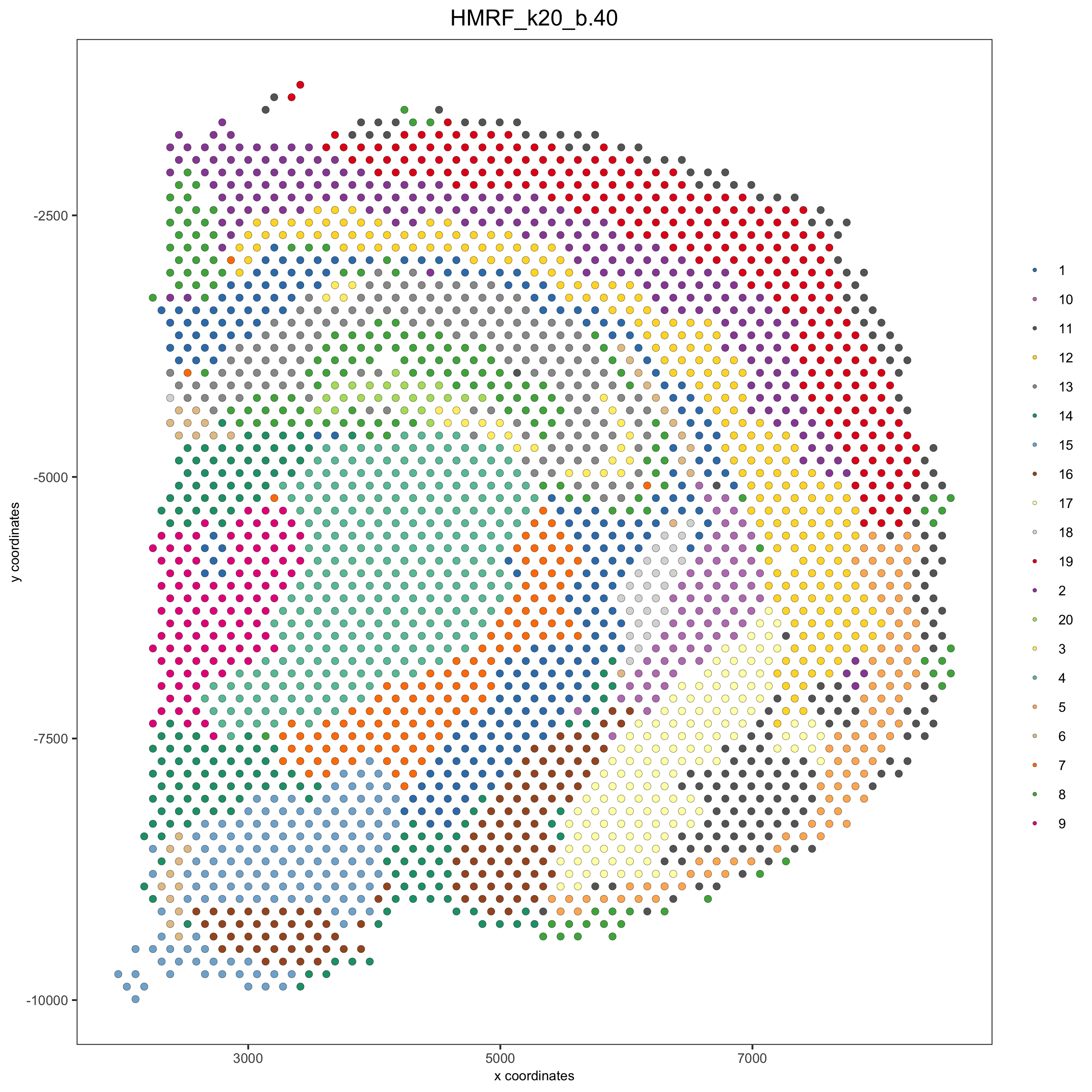
visium_brain = addHMRF(gobject = visium_brain, HMRFoutput = HMRF_spatial_genes,
k = 20, betas_to_add = c(0, 10, 20, 30, 40),
hmrf_name = 'HMRF')
spatPlot(gobject = visium_brain, cell_color = 'HMRF_k20_b.40',
point_size = 2, save_param=c(save_name="10_d_spatPlot2D_HMRF"))
11. Export and Create Giotto Viewer¶
# check which annotations are available
combineMetadata(visium_brain, spat_enr_names = 'PAGE')
# select annotations, reductions and expression values to view in Giotto Viewer
viewer_folder = paste0(results_folder, '/', 'mouse_Visium_brain_viewer')
exportGiottoViewer(gobject = visium_brain,
output_directory = viewer_folder,
spat_enr_names = 'PAGE',
factor_annotations = c('in_tissue',
'leiden_clus',
'HMRF_k20_b.40'),
numeric_annotations = c('nr_genes',
'clus_25'),
dim_reductions = c('tsne', 'umap'),
dim_reduction_names = c('tsne', 'umap'),
expression_values = 'scaled',
expression_rounding = 2,
overwrite_dir = T)
save_param = list(save_name = '6_e_metaheatmap_scran'))